Ultimo aggiornamento 2022-09-22 14:18:14
SINDROME DA DEFIBRINAZIONE o COAGULAZIONE INTRAVASCOLARE DISSEMINATA (CID)
La coagulopatia più importante dal punto di vista ostetrico-ginecologico è la cosiddetta sindrome da defibrinazione o CID, in cui si realizza un consumo o una distruzione della fibrina e del fibrinogeno circolante con incoagulabilità finale del sangue. Può avere evoluzione acuta con un quadro clinico drammatico con emorragie profuse e incoagulabilità del sangue secondaria a riduzione del fibrinogeno circolante e lisi accelerata dei coaguli. Invece, altre volte, il processo di defibrinazione si svolge in modo subacuto o cronico, di modo che i fenomeni emorragici sono scarsi o assenti. La CID subacuta può associarsi a complicanze tromboemboliche quando si verifichino situazioni di ipercoagulabilità che comprendono la trombosi venosa profonda, vegetazioni trombotiche della valvola aortica ed emboli arteriosi che prendano origine da tali vegetazioni. In tali situazioni è difficile che si verifichi un sanguinamento anomalo mentre sono frequenti i fenomeni di necrosi tissutale. Una terza manifestazione clinica della sindrome da defibrinazione è la fibrinolisi patologica o proteolisi patologica o, con sigla inglese, P.F. (Pathologic Fibrinolysis). In tale evenienza, fin dall’inizio, vi è un’abnorme conversione di plasminogeno in plasmina a causa di un deficit secretivo o strutturale del PAI-1 (Plasminogen Activator Inhibiting-1). La plasmina provoca lisi della fibrina già formata ed anche del fibrinogeno circolante e di molti altri fattori proteici della coagulazione con incoagulabilità del sangue senza fase trombotica.

ETIOLOGIA: Gli stati morbosi più comuni che possono scatenare ad una sindrome da defibrinazione possono essere così elencati:
- shock (post-emorragico, anafilattico, ecc.): probabilmente a causa della generazione di attività di fattore tissutale (TF) su monociti e cellule endoteliali
- distacco di placenta normalmente inserita;
- ritenzione prolungata di feto morto;
- aborto settico
- setticemia soprattutto da germi gram negativi la cui endotossina causa generazione di attività di fattore tissutale sulla membrana citoplasmatica dei monociti e delle cellule subendoteliali.
- embolia di liquido amniotico;
- gestosi;
- Trombofilia ereditaria o secondaria
- Trombosi venosa profonda
- Protesi valvolari cardiache
- reazioni emolitiche post-trasfusionali in caso di incompatibilità del gruppo sanguigno
- leucemia;
- porpora fulminante;
- sindrome di Waterhouse-Friderichsen: coagulopatia da consumo con emorragia cutanea, shock, rigidità nucale ed emorragie cutanee e surrenali nella sepsi da meningococco:
- Veleno di serpenti con ingresso in circolo di enzimi che attivano il fattore X o la protrombina o che convertono direttamente il fibrinogeno in fibrina.
- stati neoplastici terminali: DIC cronica dove la presenza della neoplasia può condurre a condizioni di trombofilia o a diatesi emorragica a causa della liberazione in circolo di sostanze tromboplastinosimili.
FISIOPATOLOGIA DELLA CID: I meccanismi con cui si realizza la defibrinazione sono due: la coagulazione intravascolare e l’attivazione primaria del sistema fibrinolitico.
1) la coagulazione intravale è il meccanismo più frequente; questa forma è nota anche con il nome di coagulopatia da consumo oppure CID-coagulazione intravascolare disseminata o infine in lingua inglese con la sigla D.I.C. (Disseminated Intravascular Coagulation).  Essa è innescata dalla penetrazione in circolo di fattori tromboplastinici come il Fattore tissutale (Tissue Factor o TF o Fattore III o CD142) liberato dal tessuto sottoendoteliale (miocellule, fibroblasti), dalle piastrine e dai monociti.
Essa è innescata dalla penetrazione in circolo di fattori tromboplastinici come il Fattore tissutale (Tissue Factor o TF o Fattore III o CD142) liberato dal tessuto sottoendoteliale (miocellule, fibroblasti), dalle piastrine e dai monociti.
Il Fattore tissutale (TF) è una glicoproteina integrale di membrana, di 47 KDa, costituita da 263 aminoacidi, espressa dalle cellule che in condizioni normali non sono esposte al passaggio del sangue (14-16). Quando i vasi sanguigni si rompono in modo traumatico o in seguito a rottura di placche ateromasiche, l’esposizione di cellule che esprimono il Fattore tissutale permette la complessa interazione con il fattore VII che viene attivato in VIIa (14-18). La superficie interna del vaso sanguigno è costituita da cellule endoteliali, che non esprimono il Fattore tissutale, eccetto quando vengono esposte a molecole infiammatorie come il tumor necrosis factor-alpha (TFN) (18). Un altro tipo cellulare che esprime il Fattore tissutale sulla sua superficie in condizioni infammatorie è il monocita (19-22). Anche l’attivazione primaria piastrinica induce l’espressione del TF cellulare e dalla superficie dei monociti (19-25).
Il TFN innesca la cascata coagulatoria che coinvolge il fattore VII° e gli ioni calcio (Ca++) ha come principale tappa la conversione della protrombina circolante a trombina che causa la precipitazione del fibrinogeno sotto forma di monomeri di fibrina che avvolgono i coaguli rossi piastrinici stabilizzandoli (coaguli bianchi o trombi stabilizzati). Il meccanismo coagulatorio così descritto è detto coagulazione per via estrinseca ed i fattori tutti che partecipano ad essa costituiscono la tromboplastina estrinseca. Ma esiste anche una via intrinseca della coagulazione innescata da lesioni vascolari, emorragie massive, veleni, endotossine, liquido amniotico e fosfolipidi. Interessa i fattori IXa. VIIIa, Ca++ che costituiscono la tromboplastina intrinseca. Sia la tromboplastina intrinseca che quella estrinseca agiscono infine sulla formazione di trombina tramite l’attivazione del fattore X in Xa.
Quasi subito, la presenza di tali microtrombi attiva localmente i processi fibrinolitici che attaccano enzimaticamente la fibrina, il fibrinogeno circolante residuo e molti altri fattori proteici della coagulazione allo scopo di ripristinare la pervietà vascolare. Il succedersi continuo e grave di questo processo comporta l’esaurimento del fibrinogeno, della fibrina, delle piastrine e dei fattori della coagulazione e conseguente incoagulabilità del sangue ed emorragia diffusa a tutti i distretti corporei come gengive, labbra, peritoneo, endometrio, etc.
I prodotti di degradazione della fibrina e del fibrinogeno che si formano come risultato dell’azione fibrinolitica, sono noti con la sigla P.D.F. (Products Degratation Fibrin) e, fra di essi, in particolar i D-dimeri. I PDF sono sostanze 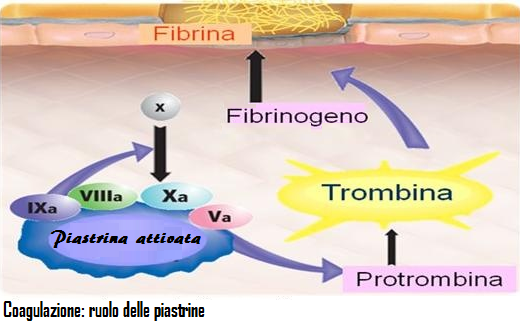 solubili, capaci di esercitare un’azione antitrombinica, anticoagulante inibendo la polimerizzazione dei monomeri di fibrina e l’azione trombizzante delle piastrine. Il nome D-dimero deriva dal fatto che questa sostanza è costituito da due frammenti D della fibrina (12,13).
solubili, capaci di esercitare un’azione antitrombinica, anticoagulante inibendo la polimerizzazione dei monomeri di fibrina e l’azione trombizzante delle piastrine. Il nome D-dimero deriva dal fatto che questa sostanza è costituito da due frammenti D della fibrina (12,13).
La coagulazione intravascolare disseminata è dunque caratterizzata dalla coesistenza: da un lato, di una coagulazione intravasale patologica del sangue circolante, con consumo del fibrinogeno e di altri fattori della coagulazione comprese le piastrine; dall’altro lato, di un’esaltazione locale della fibrinolisi a livello della microcircolazione periferica con immissione in circolo di P.D.F. dotati anch’essi di azione anticoagulante. Il risultato è un grave difetto emocoagulatorio con emorragia da ipofibrinogenemia e iperfibrinogenolisi secondaria aggravata dall’azione anticoagulante dei prodotti di degradazione del fibrinogeno (F.D.P).
Alcuni distinguono una prima fase di coagulazione da una seconda fase di iperfibrinolisi reattiva secondaria. In realtà i due fenomeni sono quasi sempre contemporanei, sebbene inizialmente possa prevalere il primo e, successivamente, il secondo.
2) attivazione primaria del sistema fibrinolitico: denominata anche fibrinolisi patologica o proteolisi patologica o, con sigla inglese, P.F. (Pathologic Fibrinolysis). In tale evenienza, fin dall’inizio, vi è un’abnorme conversione di plasminogeno in plasmina (enzima proteolitico) che provoca lisi della fibrina già formata ed anche del fibrinogeno circolante e di molti altri fattori proteici della coagulazione. Ne risulta, anche in questo caso, una riduzione del fibrinogeno circolante ed un’incoagulabilità del sangue. La demolizione enzimatica del fibrinogeno dà origine ai PDF dotati di azione anticoagulante. Tra i fattori eziologici della fibrinolisi patologica vi è anzitutto un deficit congenito del fattore inibente la conversione del plasminogeno in plasmina (PAI-1 e PAI-2: Plaminogen Activator Inhibitor). Il PAI 1-2 è prodotto soprattutto dalle cellule endoteliali vascolari e dalle piastrine ma anche dalle cellule muscolari lisce dei vasi e dalle cellule epatiche. Il PAI inibisce l’azione del tPA (tissue Plasminogen Activator), il più potente agente fibrinolitico presente nell’organismo e prodotto dalle cellule endoteliali vascolari e adipose. Al contrario, un’eccessiva concentrazione plasmatica di PAI costituirebbe un gravissimo fattore di rischio per patologie trombotiche.
Il deficit parziale o totale di PAI-1 è una condizione molto rara, trasmessa in maniera autosomica dominante. Le persone affette presentano uno (eterozigote) o due (omozigote) alleli mutati, rispettivamente, con deficit parziale o totale dell’antigene di PAI-1. In pochi pazienti la proteina è presente ma è funzionalmente inattiva.
Il deficit di PAI-1, qualitativo o quantitativo, causa la lisi prematura dei coaguli ematici e una lieve emorragia. Solo raramente si osservano sanguinamenti spontanei, però nelle pazienti omozigoti (deficit totale di PAI) si possono osservare emorragie alle ginocchia, ai gomiti, al naso e alle gengive anche per lievi traumi. Dopo interventi chirurgici possono presentarsi mestruazioni abbondanti o sanguinamento prolungato. Le emorragie sono meno frequenti e meno gravi negli eterozigoti (deficit parziale).
La diagnosi precoce del deficit antigenico o funzionale di PAI-1 è necessaria, perché gli episodi emorragici possano essere trattati con sicurezza e efficacia, con inibitori della fibrinolisi (acido epsilon-aminocaproico o acido tranexamico), che consentono di evitare il ricorso alle trasfusioni di sangue e agli emoderivati. La diagnosi si basa sul dosaggio antigenico (ELISA) e funzionale (test cromogenico) del PAI-1. In alcuni casi può rendersi necessaria l’analisi genotipica.
Contrariamente a quanto avviene nella coagulazione intravascolare del fibrinogeno circolante, nella fibrinolisi patologica non si verifica blocco della microcircolazione ed è molto rara.
CLINICA
Il quadro clinico, sia nella CID che nella P.F., dipende dall’entità del disturbo emocoagulatorio e della velocità con cui esso si instaura.
Nella CID subacuta i rilievi sono rappresentati da trombocitopenia, tempo di protrombina normale (PT) normale o appena prolungato, un tempo di tromboplastina parziale (PTT) corto, fibrinogenemia normale o lievemente ridotta e aumentato tasso dei prodotti di degradazione della fibrina. Poiché in queste affezioni vi è di solito un’aumentata produzione di fibrinogeno, è da considerare anomalo in questi pazienti il rilievo di una fibrinogenemia ai limiti bassi del range di normalità (<175 mg/dl).
Nelle forme acute, con consumo o distruzione quasi completa del fibrinogeno circolante, si osservano emorragie profuse dall’utero, dalle eventuali lacerazioni del canale del parto o dalle ferite chirurgiche; vi possono essere inoltre emorragie cutanee sotto forma di vaste zone ecchimotiche ed emorragie delle mucose: gengivorragie, epistassi, ematemesi (vomito con presenza massiva di sangue), melena, dispnea da versamento pleurico. La paziente presenta shock, con polso piccolo e frequente, agitazione, pallore, sudorazione fredda e labbra cianotiche, cianosi ingravescente, freddezza delle dita delle mani e dei piedi, ematuria, lacrime miste a sangue, insufficienza cardio-respiratoria acuta, fino al coma irreversibile ed all’exitus. Anche il sangue perso dalla donna non è mescolato a coaguli e non coagula sui teli o nei recipienti di raccolta.
Vi sono anche i sintomi secondari all’occlusione della microcircolazione periferica, secondari ma non meno gravi. A seconda dei distretti interessati e dell’estensione del fenomeno, possono manifestarsi sintomi neurologici (coma, delirio), gastrointestinali (formazione di ulcere), epatici (ittero, necrosi parenchimale), renali (necrosi corticale con oliguria o anuria). I depositi di fibrina possono causare anche un danno meccanico ai globuli rossi.
Esami di laboratorio:
SEGNI PRECOCI o TROMBOTICI
– monomeri di fibrina (fibropeptide A e B): elevate concentrazioni plasmatiche sono caratteristiche precoci della prima fase della CID
– trombocitopenia molto marcata (<100.000/mm3); segno precoce di CID
– AT III (Antitrombina III): ridotta e conseguente mancata o ridotta azione inibente sulla attivazione del fattore X in Xa che trasforma la protrombina in trombina.
– dimnuzione della proteina C attivata: è un anticoagulante naturale inibendo la conversione del fattore X in Xa
SEGNI DI FIBRINOLISI:
– ipofibrinogenemia: <300 mg/ml (v.n. 400 mg/ml)
– presenza in circolo di prodotti di degradazione della fibrina: PDF (>40 µg/ml; v.n. <10 µg/ml) e D-Dimeri (>2.000 ng/ml; v.n. 0-250 ng/ml). La misurazione presenta alta sensibilità ma bassa specificità, pertanto un valore basso può escludere una patologia trombo-embolica, ma un valore elevato non è sufficiente a determinarne la diagnosi. In conclusione: se le concentrazioni plasmatiche dei D-dimeri sono nella norma è assolutamente da escludere una CID in fase acuta (12,13). Questo perchè quando la trombina trasforma il fibrinogeno in fibrina, attiva anche il fattore XIII alla formazione di una transglutaminasi plasmatica, il fattore XIIIa che a sua volta stabilizza la fibrina legando in modo crociato le catene gamma del fibrinogeno nella zona del gruppo D. La lisi, operata dalla plasmina (o fibrinolisina), della fibrina legata in modo incrociato al fattore XIIIa, dà luogo ai prodotti di degradazione della fibrina che contiene la parte con legami crociati denominata gruppo del dimero D. Il ritrovamento nel plasma di derivati della fibrina con legami crociati, conferma la formazione della trombina e l’attivazione del fattore XIII con fibrinolisi reattiva.
– PT (tempo di protrombina) marcatamente prolungato (v.n. 10-13”): Il tempo di protrombina o tempo di Quick, noto anche come PT (sigla dall’inglese prothrombin time) e le sue misure derivate (protrombina ratio o PR e rapporto internazionale normalizzato o INR, International Normalized Ratio: v.n. 0.8-1.2) sono misure della via estrinseca della coagulazione (1-3).
Possibili cause di prolungamento dell’INR sono:
- terapia anticoagulante con antagonisti della vitamina K (dicumarolici come il warfarin)
- deficit di vitamina K.
- grave insufficienza epatica.
- trasfusioni.
- CID (Coagulazione Intravascolare Disseminata) .
– a-PTT (tempo di tromboplastina parziale: il tempo che intercorre tra l’aggiunta di calcio + tromboplastina parziale e la formazione del coagulo di fibrina all’interno dello stesso) marcatamente prolungato (4-11).
– Riduzione del fattore V e VIII, che vengono inattivati poiché in corso di CID si forma proteina C attivata.
– Riduzione del fattore VII: Il fattore VII è una proteina plasmatica glicosilata che viene sintetizzata principalmente nel fegato e che circola nel plasma sotto forma di fattore VII attivato (1%) o sotto forma di zimogeno (99%) privo di attività catalitica.
– aumento della proteina C attivata
– Transaminasi elevate conseguentemente a necrosi tissutale epatica da trombosi microvascolare

Tromboplastina o Fattore III o Fattore tissutale: termine desueto che designa  i due complessi che durante la coagulazione attivano il fattore X per iniziare la via finale comune. I due complessi sono la tromboplastina intrinseca (fattore IXa, fattore VIIIa, ione calcio e fosfolipidi) e quella estrinseca (fattore VIIa e tessutale). Il Fattore tissutale è espresso dalle cellule che in condizioni normali non sono esposte al passaggio del sangue Come le cellule sub-endoteliali e le miocellule vascolari e i fibrobalsti. Quando i vasi sanguigni si rompono, l’esposizione di cellule che esprimono il fattore tissutale permette la complessa interazione con il fattore VII, che viene attivato.
i due complessi che durante la coagulazione attivano il fattore X per iniziare la via finale comune. I due complessi sono la tromboplastina intrinseca (fattore IXa, fattore VIIIa, ione calcio e fosfolipidi) e quella estrinseca (fattore VIIa e tessutale). Il Fattore tissutale è espresso dalle cellule che in condizioni normali non sono esposte al passaggio del sangue Come le cellule sub-endoteliali e le miocellule vascolari e i fibrobalsti. Quando i vasi sanguigni si rompono, l’esposizione di cellule che esprimono il fattore tissutale permette la complessa interazione con il fattore VII, che viene attivato.
![]()
SHOCK EMORRAGICO e CID:
SEGNI INIZIALI DI SHOCK: Una riduzione della volemia comporta l’immediata attivazione dei barocettori situati a livello della carotide, dell’arco aortico, dell’atrio sinistro e delle vene polmonari. Tale attivazione conduce, attraverso un arco riflesso, ad una riduzione del tono vagale, ad un aumento della secrezione di Noradrenalina (aumento del tono del sistema simpatico) a livello delle terminazioni nervose (in particolare a livello del cuore e dei vasi sanguigni) e ad un aumento della secrezione di Adrenalina e Noradrenalina nel torrente ematico per opera della Midollare della Ghiandola Surrenale. A livello cardiaco, la riduzione di attività del sistema nervoso parasimpatico e
l’aumentato rilascio di Noradrenalina provocano tachicardia ed aumento della forza di contrazione cardiaca. A livello vascolare, la Noradrenalina determina un aumento della contrazione degli sfinteri
precapillari (prevalentemente dei vasi splancnici e muscoloscheletrici) ed un conseguente e consensuale aumento delle resistenze periferiche che costituiscono l’elemento determinante il valore della Pressione Diastolica. Dunque, le prime manifestazioni dell’ipovolemia sono in genere rappresentate da tachicardia e da moderato aumento della pressione diastolica con conseguente riduzione dei valori della pressione differenziale. In accordo alla massiva scarica simpato-adrenergica il paziente presenta inoltre agitazione ed apprensione, cute pallida e fredda, sudorazione algida e piloerezione.
La microcircolazione è composta da arteriole (“vasi di resistenza”), capillari e venule. Le arteriole sono denominate vasi di resistenza in quanto sono dotate di un manicotto di cellule muscolari liscie (sfintere precapillare) che costituisce la porta di ingresso che regola il flusso fra arterie e rete capillare. Esse inoltre sono in relazione diretta con le venule attraverso diramazioni (anastomosi artero-venose) la cui apertura consente il passaggio diretto del sangue dalle arteriole alle venule saltando il letto capillare. I capillari mediante una complessa rete di anastomosi confluiscono in una venula comune che raccoglie il sangue refluo dai tessuti. Il diametro dei capillari è regolato dalla costrizione o rilasciamento della muscolatura liscia vascolare ed è dipendente dall’attività metabolica (locale) dei tessuti, dall’attività dei nervi vascolari (attività nervosa simpatica) e dall’attività degli ormoni circolanti. Nelle prime fasi dello shock l’iperattività simpatica determina un aumento del tono vasomotore e una riduzione della capacità venosa finalizzate a garantire un riempimento atriale adeguato. L’aumento delle resistenze periferiche si realizza grazie alla costrizione dei vasi di resistenza precapillari. La contrazione degli sfinteri precapillari determina una riduzione della pressione idrostatica intracapillare che favorisce il richiamo di liquidi dall’interstizio. Quando la perfusione dei tessuti scende sotto un livello tale da non consentire più i processi metabolici necessari al mantenimento dell’integrità cellulare (vedi fasi finali dello shock), si assiste ad alterazioni responsabili della spirale irreversibile dello shock. L’acidosi metabolica, conseguente all’attivazione del metabolismo anaerobico innescato dalla mancanza di Ossigeno, porta ad un danneggiamento delle cellule con successivo rilascio di metaboliti vasoattivi. A livello del microcircolo si assiste ad una progressiva paralisi degli sfinteri precapillari che vanno incontro ad una progressiva dilatazione. Le venule invece mantengono il loro tono determinando un ristagno di sangue nei letti capillari (cosiddetta fase ischemica dello shock) che da un lato aggrava ulteriormente lo stato di ipovolemia, dall’altro favorisce l’aggregabilità dei globuli rossi e l’attivazione della Coagulazione Intravascolare Disseminata (CID). Questo stato favorisce l’instaurarsi della condizione clinica della sindrome multiorgano.
SEGNI DELLA FASE INTERMEDIA DELLO SHOCK:
– LATTATI: aumento della concentrazione ematica dei lattati > 4 mMol/l: la diminuzione del flusso di sangue ai tessuti degli organi non nobili (cute, muscoli, tessuto adiposo, visceri) e la consensuale carenza di ossigeno comporta, a livello cellulare, uno spostamento nella produzione di energia dal metabolismo aerobio al metabolismo anaerobio. Si viene così a creare un’aumentata produzione di Acido Lattico che prima si accumula nelle cellule e poi diffonde nel torrente sanguigno.
– DIMINUZIONE BE < -5 mEq/l: Il cambiamento dell’equilibrio Acido/Base precede qualsiasi significativa riduzione della Gittata Cardiaca. In accordo con queste variazioni decresce precocemente l’Eccesso di Basi (BE < -5 mEq/l), poiché la neutralizzazione degli H+ che si verifica in seguito alla aumentata concentrazione ematica di Acido Lattico porta ad un consumo delle Basi. Proprio in base alla riduzione del BE (v.n.−2 a +2 mmol/L), che pertanto rappresenta uno dei dati di laboratorio che indica ipoperfusione tissutale, è possibile delineare la soglia tra semplice emorragia e shock ipovolemico.
– pH e PaCO2: l’emogasanalisi arteriosa, nota semplicemente anche come emogasanalisi, emogas o EGA, è un esame che permette di misurare le pressioni parziali dei gas arteriosi (a. femorale o radiale) e il pH del sangue. L’emogasanalisi è indispensabile per la diagnosi di insufficienza respiratoria, per valutarne la gravità e seguirne il decorso durante la terapia. In questa fase il pH permane all’interno del suo range di normalità. Anche i chemocettori del SNC infatti rispondono all’acidemia, innescando un aumento della ventilazione/ minuto che si traduce in una riduzione della PaCO2 (v.n. 35-45 mm Hg o 4.7-5.9 kPa) (26).
– Il livello di coscienza va incontro a progressiva alterazione con presenza di lieve stato confusionale.
MANIFESTAZIONI DELLA III FASE DELLO SHOCK EMORRAGICO:
Dopo una perdita acuta di circa 1/3 del volume di sangue i riflessi cardiovascolari non sono più in grado di adeguare il riempimento del circuito arterioso. A livello del cuore la progressiva diminuzione del precarico compromette il riempimento atriale e provoca una riduzione della gittata cardiaca e la successiva caduta dei valori di PAS. Si assiste così al manifestarsi di uno stato di ipotensione arteriosa franca (durata >20 min.). Con il presentarsi dell’ipotensione, il paziente può non iperventilare sufficientemente a lungo per mantenere un pH arterioso normale; sopraggiunge così l’acidemia (pH < 7,38). Si ha l’attivazione dell’asse ipotalamo-ipofisi-surrene, con conseguente rilascio degli ormoni dello stress (Adrenalina e Noradrenalina) per opera della Midollare del Surrene, di Ormoni Glicocorticoidi per opera della Corticale del Surrene e di Glucagone per opera del Pancreas. Questi ormoni determinano glicogenolisi, lipolisi e lieve ipopotassiemia. Dunque, nel reparto di Emergenza, i pazienti con emorragia possono presentare lieve iperglicemia (150-170 mg/dl) ed ipokaliemia (3,5- 3,7 mEq / L). La diuresi, che ovviamente rappresenta un ottimo indicatore dello stato di ipoperfusione degli organi, si riduce severamente (< 0,5 ml / Kg / h).
L’ipotensione, pur alterando significativamente il rapporto Ventilazione/Perfusione, raramente è capace di produrre ipossiemia se le vie aeree sono pervie, il polmone non è danneggiato e lo sforzo respiratorio è adeguato.
MANIFESTAZIONI FINALI DELLO SHOCK EMORRAGICO:
A questo punto, se la perfusione tissutale è restaurata si può assistere ad un recupero; altrimenti, se l’ipovolemia persiste, la vasocostrizione periferica costituisce un fattore aggravante in quanto instaura un circolo vizioso innescato dall’ipoperfusione e dall’ampio danno cellulare. La riduzione del flusso ai centri midollari vasomotori deprime l’attività dei riflessi compensatori. Il risultato dell’ipoperfusione tissutale e del metabolismo anaerobio è quindi costituito da anossia, ipercapnia e acidosi lattica. Le riserve energetiche delle cellule diminuiscono progressivamente e, poiché l’integrità delle membrane cellulari risulta compromessa, si assiste alla liberazione nel circolo sanguigno dei componenti cellulari. La funzione cardiaca è depressa a causa del rilascio di fattori “deprimenti” (citochine) prodotti dai leucociti a livello di tutti gli organi ipoperfusi. Nel fegato, il danno da infiammazione e da Radicali liberi dell’Ossigeno prodotti dai Neutrofili è costituito da una persistente microischemia epatica, con conseguente produzione di irregolari danni centrolobulari che potrebbero portare ad un immediato aumento delle Transaminasi. A livello renale, in relazione al grado di insulto di tipo ipotensivo, si potrebbe manifestare spasmo acuto delle arteriole preglomerulari e conseguente Necrosi Tubulare Acuta con rapida evoluzione verso l’Insufficienza Renale Acuta.
Nello stato di shock conclamato si aggiunge inoltre un elemento aggravante l’ipoperfusione periferica: l’attivazione della cascata coagulativa sottoforma di Coagulazione Intravascolare Disseminata (CID). La CID si verifica a carico di tutti gli organi, ma la microischemia che si produce a livello dei vasi intestinali costituisce, probabilmente, una complicazione ulteriore dello scompenso circolatorio. Infatti, la rottura della barriera della mucosa favorirebbe, in base all’ipotesi più comunemente accettata, il passaggio di batteri e di tossine batteriche all’interno del torrente ematico. Cambiamenti simili si avrebbero a carico della rete capillare dei polmoni dove la comparsa di un edema interstiziale ed alveolare prelude ad una riduzione del passaggio dei gas (ARDS, Adult Distress Respiratory Syndrome). Poiché le sostanze batteriche sono dei potenti agenti vasodilatatori i meccanismi vasocostrittori, nonostante l’intensa attività simpatica, potrebbero essere inibiti e non essere in grado di sostenere ulteriormente la PA. In questa fase il paziente è in uno stato di coma.
In base alla progressione dello shock ipovolemico appena descritta è possibile identificare le seguenti condizioni:
- Stato di semplice emorragia o ipovolemia (shock ipovolemico leggero): Sanguinamento sospetto o perdita sospetta con FC < o > 100 / min., FR normale, PA normale, BE normale.
- Stato di emorragia o ipovolemia con ipoperfusione (shock ipovolemico moderato): Sanguinamento sospetto o perdita sospetta con BE < -5 mEq/l e persistente FC > 100 /min.
- Shock ipovolemico (shock ipovolemico grave): Sanguinamento o perdita sospetta associati ad almeno 4 tra i criteri visti.
![]()
TERAPIA DELLA CID: l’end-point primario è la risoluzione della causa della CID, mentre gli end-point clinici prevedono protocolli articolati in relazione alla presenza e gravità dei fattori trombotici o fibrinolitici. La terapia presuppone:
- – la rimozione del fattore iniziale che ha scatenato la coagulopatia: bloccare le fonti emorragiche, trattare l’infezione che la sottende, rimuovere i residui placentari nella ritenzione di placenta, rimuovere il tessuto neoplastico, aspirare le raccolte di sangue dopo gli interventi chirurgici. La rimozione del fattore eziologico iniziale è quasi sempre sufficiente a risolvere la condizione.
- – il trattamento specifico del disordine emocoagulatorio;
- – la reintegrazione della volemia.
Quando si manifesta il quadro acuto della defibrinazione, il trattamento del disordine emocoagulatorio può essere eseguito in modo razionale solo nei pochissimi casi in cui è possibile discriminare fra C.I.D. e P.F. e, nell’ambito della C.I.D., fra fase in cui prevale la deposizione di microtrombi e la fase di fibrinolisi reattiva.
L’eparina è il farmaco da utilizzare in prima istanza nella CID subacuta, negli stadi iniziali della CID, per arrestare la formazione di microtrombi ed il consumo di fibrinogeno. L’eparina, infatti, inibisce l’azione della trombina sul fibrinogeno, la generazione della tromboplastina, l’azione della tromboplastina già formata e la trasformazione della protrombina in trombina. ciò è valido solo se non sono già presenti perdite ematiche e la DIC è stata scoperta con gli opportuni esami di laboratorio ed è perciò ancora asintomatica.
L’uso di farmaci antifibrinolitici è indicato solo in un secondo tempo od un presenza di sanguinamenti clinicamente pericolosi allo scopo di ridurre un’eccessiva produzione di prodotti di degradazione della fibrina che possono prolungare lo stato emorragico.
Nella P.F., ma solo nella sua forma pura, è indicato invece l’uso di farmaci antifibrinolitici fin dall’inizio.
EPARINA: L’uso di eparina nella D.I.C., o di antifibrinolitici nella P.F. (pathologic fibrinogenolysis), deve precedere o quanto meno accompagnare la terapia trasfusionale intesa a reintegrare la volemia ed il patrimonio dei fattori della coagulazione. Senza eparina e antifibrinolitici, con la terapia trasfusionale non si fa altro che fornire nuovo substrato alla coagulopatia e, in definitiva, accentuarne ulteriormente la gravità. Come fluidi da infusione è consigliabile limitarsi al plasma o al sangue intero; le soluzioni di fibrinogeno sono da evitare anche per l’elevato rischio di trasmettere un’epatite virale e per l’elevato contenuto di plasminogeno. E’ anche preferibile evitare l’uso di sostituti plasmatici che possono accentuare la diluizione dei fattori della coagulazione o, addirittura, aggravare la coagulopatia formando complessi inattivi col fibrinogeno come nel caso del destrano.
Per quanto riguarda l’eparina, uno degli schemi di dosaggio proposto prevede la somministrazione endovenosa iniziale rapida di 50 UI/Kg di peso corporeo, seguita dalla infusione endovenosa continua di 1000 UI/ora.
Concentrati di piastrine: per correggere la trombocitopenia (e anche come fonte del fattore V piastrinico);
Crioprecipitati per reintegrare il fibrinogeno e il fattore VIII; il crioprecipitato è un emoderivato che si ottiene per separazione dal plasma congelato e sottoposto a lentissimo scongelamento. E’ formato soprattutto da fibrinogeno, dal fattore VIII della coagulazione, dalla sieroalbumina e da altri fattori della coagulazione, che possono essere utilizzati nel trattamento di particolari sindromi emorragiche (CID, afibrogenemia, emofilia A ecc.).
Plasma fresco congelato: per aumentare il tasso del fattore V e di altri fattori della coagulazione e come fonte di antitrombina III, che può anch’essa risultare ridotta a seguito di una CID.
Per quanto riguarda i farmaci antifibrinolitici, il dosaggio varia da sostanza a sostanza. L’acido tranexamico si somministra per via endovenosa (1 g ogni sei ore) e successivamente si può continuare con iniezioni endomuscolari o per via orale. Per l’acido epsilonaminocaproico (EACA), la dose iniziale è di 4-5 g per via endovenosa, seguita da 1 g ogni 4 ore, sempre per via endovenosa.
Una posizione a parte spetta ad un inibitore polipeptidico delle proteasi, estratto dalla parotide e dal polmone dei bovini noto col nome di aprotinina (Trasylol fl e.v. 100.000, 200.000, 500.000 UI). Secondo l’ipotesi oggi prevalente (peraltro non condivisa da tutti), il Trasylol presenta infatti la caratteristica di avere una doppia azione, antifibrinolitica ed antitromboplastinica; inoltre ha un periodo di emivita relativamente breve (150 minuti), fatto che riduce il rischio di fenomeni da accumulo. Il Trasylol, infine, inibisce l’attivazione delle chinine, riducendo la vasocostrizione periferica e migliorando le condizioni del microcircolo. La dose media del Trasylol è 200.000 UI per iniezione endovenosa lenta come prima somministrazione, seguita poi da infusione endovenosa alla velocità di circa 100.000 UI all’ora per un totale di 500.000-600.000 U.I.. Molti propongono di trattare con eparina tutte indistintamente le sindromi acute da defibrinazione. Altri invece sostengono che sia preferibile usare il Trasylol al posto dell’eparina, rammentando appunto che il Trasylol possiede una doppia azione antitromboplastinica ed antifibrinolitica, e quindi sarebbe in grado di contrastare efficacemente sia la tendenza alla coagulazione intravascolare sia l’attivazione, primaria o secondaria, della fibrinolisi.
– Proteina C ricombinante: bolo di 100 UI/Kg seguito da infusione di 50-80 UI/Kg ogni 6 ore fino a normalizzazione degli indici emostatici
– Proteina C attivata estrattiva: 5.000-10.000 UI ogni 6 ore
In sintesi la terapia della CID da emorragia prevede:
- Eparina
- Antifibrinolitici
- Concentrato di piastrine
- Crioprecipitati o fibrinogeno per correggere il deficit di Fibrinogeno (<1 gr/L) e fattore VIII
- Fattori della coagulazione
- Plasma fresco per reintegrare le perdite del fattore V e antitrombina III
- Trombomodulina ricombinante e Selenio: non ancora disponibili nella farmacopea italiana
- Nichols WL, Bowie EJ. Standardization of the prothrombin time for monitoring orally administered anticoagulant therapy with use of the international normalized ratio system. Mayo Clin Proc. 1993 Sep;68(9):897-8. PMID: 8371608
- Testa S, Morstabilini G, Fattorini A, Galli L. Discrepant sensitivity of thromboplastin reagents to clotting factor levels explored by the prothrombin time in patients on stable oral anticoagulant treatment: impact on the international normalized ratio system. Haematologica. 2002 Dec;87(12):1265-73.
- orsti J, Uppa H, Vilpo JA. Poor agreement among prothrombin time international normalized ratio methods: comparison of seven commercial reagents. Clin Chem. 2005 Mar;51(3):553-60. Epub 2005 Jan 21. PMID: 15665046
- Fritsma, George A. “Evaluation of Hemostasis.” Hematology: Clinical Principles and Applications . Ed. Bernadette Rodak. WB Saunders Company: Philadelphia, 2002. 719-53.
- ^ Kitchen S, Preston FE. Standardization of prothrombin time for laboratory control of oral anticoagulant therapy. Semin Thromb Hemost. 1999;25(1):17-25. PMID: 10327216
- ^ Kazama M. Quality control of ISI/INR system in oral anticoagulant therapy. [Article in Japanese] Rinsho Ketsueki. 1990 Jun;31(6):769-75. PMID: 2214167
- ^ Van den Besselaar AM. International standardization of laboratory control of oral anticoagulant therapy: a survey of thromboplastin reagents used for prothrombin time testing. J Heart Valve Dis. 1993 Jan;2(1):42-52. Review. PMID: 8269109
- ^ Opartkiattikul N. Standardization of coagulation tests. Southeast Asian J Trop Med Public Health. 1999;30 Suppl 3:79-85. PMID: 10926265
- ^ Hirsh J, Poller L. The international normalized ratio. A guide to understanding and correcting its problems. Arch Intern Med. 1994 Feb 14;154(3):282-8. Review. PMID: 8297194
- ^ a b c Goodman & Gilman, The Pharmacological Basis of Therapeutics, 12ª ed., 2011, p. 865, trad. propria.
- ^ Della Valle P , Crippa L , Safa O , Tomassini L Potential failure of the International Normalized Ratio (INR) System in the monitoring of oral anticoagulation in patients with lupus anticoagulants. Ann Med Interne (Paris). 1996 Sep;147 Suppl 1:10-4. PMID: 8952752
- S. S. Adam, N. S. Key, C. S. Greenberg, D-dimer antigen: current concepts and future prospects in Blood, vol. 113, nº 13, 2008, pp. 2878–2887
- PS. Wells, DR. Anderson; M. Rodger; M. Forgie; C. Kearon; J. Dreyer; G. Kovacs; M. Mitchell; B. Lewandowski; MJ. Kovacs, Evaluation of D-dimer in the diagnosis of suspected deep-vein thrombosis. in N Engl J Med, vol. 349, nº 13, Set 2003, pp. 1227-35,
- Edgington TS, Mackman N, Brand K, Ruf W. The structural biology of expression and function of tissue factor. Thromb Haemost 1991; 66: 67-79.
- Camerer E, Kolsto AB, Prydz H. Cell biology of tissue factor, the principal initiator of blood coagulation. Thromb Res 1996; 81: 1-41.
- Fuster V, Fallon JT, Badimon JJ, Nemerson Y. The unstable atherosclerotic plaque: clinical significance and therapeutic intervention. Thromb Haemost 1997; 78: 247-55.
- Rauch U, Nemerson Y. Tissue factor, the blood, and the arterial wall. Trends Cardiovasc Med 2000; 10: 139-43.
- Wilcox JN, Smith KM, Schwartz SM, Gordon D. Localization of tissue factor in the normal vessel wall and in the atherosclerotic plaque. Proc Natl Acad Sci USA 1989; 86: 2839-43.
- Moons AH, Levi M, Peters RJ. Tissue factor and coronary artery disease. Cardiovasc Res 2002; 53: 313-25.
- Petersen LC, Freskgard P, Ezban M. Tissue-factor-dependent factor VIIa signaling. Trends Cardiovasc Med 2000; 10: 47-52.
- Hoffman M, Monroe DM, Oliver JA, Roberts HR. Factors IXa and Xa play distinct roles in tissue factor-dependent initiation of coagulation. Blood 1995; 86: 1794-801.
- Camera M, Brambilla M, Frigerio M, et al. L’attivazione piastrinica induce l’espressione di tissue factor immunoreattivo (irTF) sulla superficie cellulare. (abstr) In: Abstracts Congresso Nazionale della Società Italiana per lo Studio dell’Aterosclerosi. Chieti, 2002: C26.
-
Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. Vastatins inhibit tissue factor in human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol 1997; 17: 265-72.
-
Soejima H, Ogawa H, Yasue H, et al. Angiotensin-converting enzyme inhibition reduces monocyte chemoattractant protein-1 and tissue factor levels in patients with myocardial infarction. J Am Coll Cardiol 1999; 34: 983-8.
-
Serena Del Turco, Raffaele De Caterina: Biologia e fisiopatologia del fattore tissutale e sua rilevanza per la patologia cardiovascolare. Ital Heart J Suppl 2003; 4 (7): 559-568
-
Harrison, Principi di medicina interna, 16ª ed., Milano, McGraw-Hill, 2005, ISBN 88-386-2999-4.
12 commenti
Hurrah! In the end I got a blog from where I be capable of in fact get valuable data regarding my study and
knowledge.
Thank you, I have recently been searching for information about this topic for a long time and yours is the best
I have discovered till now. However, what about the bottom line?
Are you positive about the source?
I every time emailed this web site post page to all my contacts, as if
like to read it next my links will too.
Thank you, I’ve just been searching for info about this subject for ages and yours is
the greatest I have found out so far. However, what concerning the conclusion? Are you positive about the source?
I think this is among the most important info for me.
And i’m glad reading your article. But wanna remark on few general things,
The website style is ideal, the articles is really nice : D.
Good job, cheers
Hello there! This article could not be written much
better! Looking at this article reminds me of my
previous roommate! He continually kept talking about this.
I most certainly will send this article to him. Fairly certain he’s going to have a great read.
Thank you for sharing!
I visited multiple blogs except the audio feature for audio songs current at this website is actually fabulous.
Neat blog! Is your theme custom made or did you download it from somewhere?
A theme like yours with a few simple adjustements would really make my blog stand out.
Please let me know where you got your theme. Cheers
After looking into a handful of the blog articles on your site, I seriously appreciate your way of blogging.
I book-marked it to my bookmark website list and will be checking back in the near future.
Please check out my web site too and tell me what you think.
I am regular reader, how are you everybody? This paragraph posted at this web site is genuinely pleasant.
Hi, every time i used to check website posts here in the early hours in the morning,
for the reason that i love to learn more and more.
If some one wishes expert view regarding running a blog then i suggest him/her to pay a quick visit this webpage, Keep up the good work.