
L’ernia diaframmatica congenita è un difetto di chiusura del muscolo diaframmatico con erniazione in cavità toracica dei visceri addominali che comprimono il polmone omolaterale e spesso anche quello controlaterale impedendone lo sviluppo. Il diaframma è una membrana muscolo-tendinea che separa la cavità toracica, in cui sono contenuti il cuore ed i polmoni, dalla cavità addominale, dove si trovano organi quali lo stomaco, il fegato, la milza e l’intestino (1.2). Nelle prime sei settimane di gravidanza esiste una comunicazione fisiologica tra addome e torace. Normalmente questa comunicazione si chiude entro il 3° mese di gestazione; la mancata fusione della membrana pleuroperitoneale con gli altri 3 componenti diaframmatici (setto trasverso, mesentere dorsale dell’esofago e parete del corpo) è causa dell’ernia diaframmatica congenita (CDH) con invasione toracica dei visceri addominali fetali (1,2). Raramente si osservano casi di CDH ad insorgenza tardiva (3,4).
esiste una comunicazione fisiologica tra addome e torace. Normalmente questa comunicazione si chiude entro il 3° mese di gestazione; la mancata fusione della membrana pleuroperitoneale con gli altri 3 componenti diaframmatici (setto trasverso, mesentere dorsale dell’esofago e parete del corpo) è causa dell’ernia diaframmatica congenita (CDH) con invasione toracica dei visceri addominali fetali (1,2). Raramente si osservano casi di CDH ad insorgenza tardiva (3,4).
Frequenza: 1/3.000 gravidanze. Può presentarsi in forma isolata o in associazione ad alterazioni cromosomiche o ad anomalie di altri organi e apparati (cuore, intestino, reni). Nel 10% dei casi si associano anomalie genetiche (S. di Pallister-Killian, Fryns, Ghersoni-Baruch, WAGR, Denys-Drash, Brachman-De Lange, Donnai-Barrow, Wolf-Hirschhorn) o cromosomiche (trisomia 13 o S. di Patau, trisomia 18 o S. di Edwards, tetrasomia 9p, dup 11q23ter, del 15q24-26, del 1q41-42.12; del 8p23.1) che evidenziano l’elevata eterogeneità genetica della CHD (5).
cromosomiche o ad anomalie di altri organi e apparati (cuore, intestino, reni). Nel 10% dei casi si associano anomalie genetiche (S. di Pallister-Killian, Fryns, Ghersoni-Baruch, WAGR, Denys-Drash, Brachman-De Lange, Donnai-Barrow, Wolf-Hirschhorn) o cromosomiche (trisomia 13 o S. di Patau, trisomia 18 o S. di Edwards, tetrasomia 9p, dup 11q23ter, del 15q24-26, del 1q41-42.12; del 8p23.1) che evidenziano l’elevata eterogeneità genetica della CHD (5).
Etiologia: i fattori responsabili della mancata fusione dei foglietti diaframmatici alla fine del 1° trimestre non si conoscono. Tale patologia non è ereditaria anche se sono stati descritti alcuni casi in più elementi della stessa famiglia. I genitori che hanno avuto un bambino con un ernia diaframmatica isolato sono ad aumentato rischio di avere un altro bambino con lo stesso problema (2%).
Classificazione:
esistono due tipi di CDH: l’ernia di Bochdalek o postero-laterale e, più raramente (2%), l’ernia retrosternale di Morgagni-Larrey, più piccola e difficilmente diagnosticabile in epoca prenatale. L’ernia di Morgagni è più frequente nei feti di sesso femminile e può passare inosservata anche dopo la nascita. L’ernia in genere è monolaterale ed è più frequente a sinistra (90% dei casi) Se l’ernia interessa l’emidiaframma destro anche il rene potrebbe dislocarsi in cavità toracica. L’erniazione dei visceri addominali avviene nel II°-III° trimestre o addirittura dopo la nascita (6).
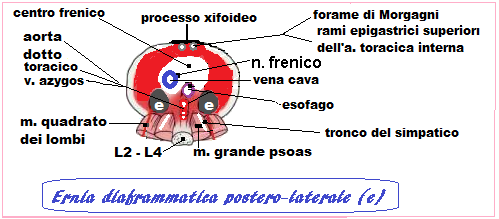
Stadiazione:
L’ernia diaframmatica congenita (CDH) è un’entità clinica in cui è possibile riconoscere diversi livelli di gravità. In casi isolati, cioè in assenza di altre anomalie fetali, la severità della patologia e di conseguenza l’outlook neonatale è determinato da tre fattori:
1. la posizione del fegato. Feti con parte del fegato erniata in torace hanno in genere una prognosi più severa.
2. il valore di LHR (lung-to-head ratio), un parametro ecografico che misura il rapporto fra l’area del polmone controlaterale al difetto erniario e la circonferenza cranica (HC) del feto. Più elevato è il valore di questo parametro, migliore è la prognosi.
3. Polidramnios: è determinato dalla compressione esercitata dai visceri addominali sull’esofago.
Feti con fegato non erniato, e un valore di LHR >1.4 (circa il 25% dei casi di ernia diaframmatica) hanno in genere una buona prognosi e pur richiedendo cure intensive neonatali, sono caratterizzati da un buon esito a breve, medio e lungo termine.
Feti con erniazione del fegato e un valore di LHR<1, sono caratterizzati da una prognosi infausta determinata da gravissima ipoplasia polmonare, ipertensione polmonare (da ipertrofia muscolare delle arterie polmonare) e morte fetale o perinatale nel 90% dei casi e quindi occorre intervenire chirurgicamente al più presto possibile (26-28a w).
Diagnosi: La diagnosi di CDH in gravidanza viene generalmente effettuata, con difficoltà, nel III° trimestre o dopo la nascita.
USG: il diaframma è visibile come una sottile linea anecogena e arcuata con convessità rivolta in alto che separa i polmoni dal fegato. Il 50% dei casi di CDH possono essere identificati durante il periodo fetale, generalmente nel III° trimestre di gravidanza, quando all’esame ecografico sia possibile evidenziarne segni diretti, quali l’erniazione dell’intestino e/o del fegato nel torace. Lo stomaco appare come un’area cistica anecogena paracardiaca che si modifica con i movimenti peristaltici. Inoltre, durante l’inspirazione fetale, queste strutture anomale osservate nel torace, si spostano verso l’addome mentre il diaframma si sposta in direzione opposta, in alto.
Segni indiretti sono rappresentati da CA inferiore alla norma (-2 DS) a causa della vacuità dell’addome, assenza di movimenti respiratori nell’emitorace sede di ernia, il polidramnios da compressione esofagea, lateralizzazione del cuore, shift sx-dx del cuore e dei grossi vasi (7-9).


Diagnosi differenziale: deve porsi rispetto a patologie primitive del torace e dei polmoni come la malformazione adenomatoide cistica congenita del polmone (CCAM), il sequestro bronco-polmonare e teratomi.
RMN: si può osservare profilo diaframmatico irregolare, parzialmente o totalmente assente, erniazione nel torace delle anse addominali, milza, fegato, dislocazione in alto di uno o entrambi i polmoni. Lateralizzazione del cuore. L’esame non richiede sedazione, dura 30′ ed è privo di controindicazioni e dolore per la madre ed il feto.
Terapia:
1) Nel 25% dei casi l’ernia diaframmatica si presenta con fegato non erniato e LRH >1.4. Il difetto erniario può essere corretto con intervento chirurgico dopo la nascita del bambino senza nessuna complicazione. la prognosi è quasi sempre buona.
2) F.E.T.O. (Fetal Endoscopic Tracheal Occlusion): nei casi di erniazione del fegato e LRH <1 è necessario intervenire il più precocemente possibili per evitare i gravissimi danni fetali prima descritti. L’intervento di F.E.T.O. blocca la fuoriuscita delle secrezioni bronchiali nel liquido amniotico. In tal modo i polmoni non vengono collassati e possono maturare ed aumentare di volume. Una sonda è
L’intervento di F.E.T.O. blocca la fuoriuscita delle secrezioni bronchiali nel liquido amniotico. In tal modo i polmoni non vengono collassati e possono maturare ed aumentare di volume. Una sonda è  introdotta nella trachea fetale alla 26-28a e rimossa alla 35a settimana. Percentuali di sopravvivenza: 50-70%. L’intervento dura circa 30′ ed è effettuata con la gravida in anestesia epidurale ed il feto sedato, per diminuirne la motilità, con tranquillante iniettato nei glutei fetali con iniezione endoscopica (4-6). L’intervento è gravato da un’alta percentuale di parto prematuro. La paziente dovrà essere sotto stretto controllo clinico e terapia tocolitica per tutta la durata della gravidanza. Il parto avverrà mediante T.C. di elezione alla 37-38a w. Il neonato dovrà essere immediatamente intubato e sottoposto a respirazione artificiale. Appena le condizioni cliniche lo permetteranno sarà sottoposto a intervento chirurgico definitivo (7,8).
introdotta nella trachea fetale alla 26-28a e rimossa alla 35a settimana. Percentuali di sopravvivenza: 50-70%. L’intervento dura circa 30′ ed è effettuata con la gravida in anestesia epidurale ed il feto sedato, per diminuirne la motilità, con tranquillante iniettato nei glutei fetali con iniezione endoscopica (4-6). L’intervento è gravato da un’alta percentuale di parto prematuro. La paziente dovrà essere sotto stretto controllo clinico e terapia tocolitica per tutta la durata della gravidanza. Il parto avverrà mediante T.C. di elezione alla 37-38a w. Il neonato dovrà essere immediatamente intubato e sottoposto a respirazione artificiale. Appena le condizioni cliniche lo permetteranno sarà sottoposto a intervento chirurgico definitivo (7,8).

3) Amnioriduzioni: allo scopo di favorire la respirazione materna, ridurre il tono delle pareti uterine, prolungare la durata della gestazione e consentire la maturazione polmonare fetale.
4) IVG: questa patologia consente l’interruzione volontaria della gravidanza entro il 180° giorno di amenorrea (Legge 194, art. 6)
Valutazione prenatale: prima di sottoporsi all’intervento e’ necessaria una valutazione prenatale completa, che preveda amniocentesi/villocentesi per l’indagine sul cariotipo fetale e un’ecografia di III° livello per escludere altre malformazioni (9).
Complicanze: L’intervento e’ in genere semplice e privo di complicazioni per la madre. Esistono tuttavia dei rischi per la gravidanza: emorragia, distacco di placenta, morte del feto, rottura prematura delle membrane e parto prematuro con percentuali del 15-30%.
L’intervento viene eseguito in Europa presso 4 centri, all’interno del programma “ Clinical Fetoscopic Endotracheal Occlusion Program”:
– University Hospital “Gasthuisberg”- Leuven , Belgio (Prof J Deprest)
-King’s College Hospital- Londra , UK (Prof K Nicolaides)
-Vall d’Hebron Hospital-Bercelona, Spagna (Prof E Gratacos)
Clinica “Mangiagalli” di Milano

Enjoy this exciting new issue and stay tuned for more!
We value your opinion and are receptive to comments and suggestions.
Yours faithfully,
dr. Enzo Volpicelli
References list:
-
Lin AE, Pober BR, Adatia I. Congenital diaphragmatic hernia and associated cardiovascular malformations: type, frequency, and impact on management. Am J Med Genet C Semin Med Genet. 2007;145C:201–216.
-
A. Giannotta, E. Chiella, D. Codrich, M. Monai, L. Paduano, G. Pelizzo, A. Messineo. ERNIA DIAFRAMMATICA CONGENITA: UN CASO DI DIAGNOSI TARDIVA.. Medico e Bambino pagine elettroniche 2001; 4(1) http://www.medicoebambino.com/?id=CH0101_10.html
-
Logan JW, Rice HE, Goldberg RN, Cotten CM. Congenital diaphragmatic hernia: a systematic review and summary of best-evidence practice strategies. J Perinatol. 2007 Sep;27(9):535-49.
- Tovar JA. Congenital diaphragmatic hernia. Orphanet J Rare Dis. 2012 Jan 3;7:1.
- Deprest J, Jani J, Van Schoubroeck D, Cannie M, Gallot D, Dymarkowski S, Fryns JP, Naulaers G, Gratacos E, Nicolaides K. Current consequences of prenatal diagnosis of congenital diaphragmatic hernia. J Pediatr Surg. 2006 Feb;41(2):423-30. Review.
- Fotter R, Schimpl G, Sorantin E, Fritz K, Landler U. Delayed presentation of congenital diaphragmatic hernia. Pediatr Radiol 1992, 22(3):187-91.
- Stokes KB. Unusual varieties of diaphragmatic herniae. Prog Pediatr Surg 1991, 27: 127-47.
- Numanoglu A, Steiner Z, Millar A, Cywes S. Delayed presentation of congenital diaphragmatic hernia. S Afr J Surg 1997, 35(2): 74-76.
- Deprest J, Jani J, Gratacos E, Vandecruys H, Naulaers G, Delgado J, Greenough A, Nicolaides K; FETO Task Group.Fetal intervention for congenital diaphragmatic hernia: the European experience. Semin Perinatol. 2005 Apr;29(2):94-103. Review.
- Jani J, Gratacos E, Greenough A, Piero JL, Benachi A, Harrison M, Nicolaides K, Deprest J; FETO Task Group.Percutaneous fetal endoscopic tracheal occlusion (FETO) for severe left-sided congenital diaphragmatic hernia. Clin Obstet Gynecol. 2005 Dec;48(4):910-22.
- Dekoninck P, Gratacos E, Van Mieghem T, Richter J, Lewi P, Ancel AM, Allegaert K, Nicolaides K, Deprest J.: “Results of fetal endoscopic tracheal occlusion for congenital diaphragmatic hernia and the set up of the randomized controlled TOTAL trial”. Early Hum Dev. 2011 Sep;87(9):619-24. doi: 10.1016/j.earlhumdev.2011.08.001.
- Ruano R, Yoshisaki CT, da Silva MM, Ceccon ME, Grasi MS, Tannuri U, Zugaib M.: “A randomized controlled trial of fetal endoscopic tracheal occlusion versus postnatal management of severe isolated congenital diaphragmatic hernia”. Ultrasound Obstet Gynecol. 2012 Jan; 39(1):20-7. Epub 2011 Dec 14.