
L’iperplasia surrenalica congenita CAH (Congenital Adrenal Hyperplasia) è il termine che insieme a sindromi adreno-genitali viene comunemente usato per descrivere un gruppo di patologie autosomiche recessive dovute alla mancanza di uno dei 5 enzimi che intervengono nella sintesi del cortisolo e aldosterone nella corteccia surrenalica (1-5). La mutazione comporta una ridotta produzione di ormoni glucocorticoidi e mineralcorticoidi, con conseguente aumento di androgeni (sindrome adreno-genitale).
Eziologia: Il difetto enzimatico più frequente è quello che interessa l’enzima 21-idrossilasi, appartenente alla famiglia dei citocromi P450 e costituito da 494 aminoacidi, che determina oltre il 90-95 % delle iperplasie surrenaliche congenite. Le mutazioni del gene CYP21, situato sul cromosoma 6, che codifica l’enzima, sono responsabili del deficit di 21-idrossilasi (6-9).
La sintesi degli ormoni steroidei surrenalici è un processo complesso che a partire dal colesterolo permette la sintesi del cortisolo, dell’aldosterone e degli ormoni sessuali. Le ghiandole endocrine che sintetizzano gli steroidi sono il corticosurrene e le gonadi. Nell’ovaio i compartimenti capaci di sintetizzare gli steroidi sono follicolo, corpo luteo e cellule interstiziali (in particolare le cellule parailari).
Il corticosurrene presenta due particolari peculiarità:
• è in grado di secernere il più ampio spettro di steroidi (glucocorticoidi, mineralcorticoidi, androgeni e, in piccole quantità, estrogeni);
• presenta zone anatomiche ben distinte (glomerulare, fascicolata e reticolata) con diverse attività, meccanismi di secrezione e di controllo da parte di altri ormoni (feed-back).
Altri organi (fegato, cute, tessuto adiposo, muscolo, ecc.) sono in grado di metabolizzare molecole steroidee, trasformandole in metaboliti più o meno attivi o completamente inattivi. Tuttavia, al contrario degli organi steroidogenetici veri e propri, questi non sono sottoposti al controllo a feed-back da parte di altri ormoni (LH, ACTH, Angiotensina II) in grado di modulare l’attività delle vie enzimatiche, modificando la secrezione in circolo di uno steroide specifico. Pertanto, questi organi sono soprattutto sede del catabolismo degli steroidi e la loro attività enzimatica è una caratteristica intrinseca che viene modulata dalla presenza e quantità dei substrati, e da fattori regolatori (ormonali e non) che agiscono con meccanismi paracrini.
L’enzima 21-idrossilasi (chiamato anche CYP21 o P450c21) appartiene alla categoria dei citocromi P-450 e, a livello intracellulare, è localizzato nel reticolo endoplasmatico. Esso catalizza la conversione del 17-idrossi progesterone (17OH-P) in 11-desossicortisolo, un precursore del cortisolo, e del progesterone in desossicorticosterone, precursore dell’aldosterone (10-15).
Frequenza – La cosiddetta forma classica, con virilizzazione e perdita di sale, si manifesta in epoca neonatale o nelle prime fasi dell’infanzia (in Italia, 1:16000 nati). La forma non classica (virilizzazione semplice e conservata secrezione di aldosterone) è più frequente e si riscontra nello 0,2 % della popolazione bianca in generale con un picco del 2% negli ebrei Ashkenaziti e negli ispanici.
Fisiopatologia – Nel caso in cui vi sia un deficit, parziale o totale, nella funzionalità dell’enzima 21-idrossilasi il paziente portatore del difetto non è in grado di sintetizzare efficientemente una adeguata quantità di cortisolo e/o aldosterone. Pertanto, i bassi valori di cortisolo prodotti esercitano un feed back positivo sull’ipotalamo e sull’ipofisi con ipersecrezione di ormone adrenocorticotropo (CRH) ipotalamico e ACTH ipofisario che a sua volta determina una iperstimolazione della corteccia corticosurrenalica (16-32).
In seguito all’iperstimolazione surrenalica si assiste ad un accumulo dei precursori del cortisolo che, nella sequenza biosintetica degli ormoni surrenalici, sono posti a monte del difetto enzimatico (17-OH-progesterone). I precursori accumulati, pertanto, non potendo proseguire lungo la via che dovrebbe portarli verso la sintesi del cortisolo, vengono deviati verso altre vie biosintetiche con iperproduzione di ormoni sessuali maschili (androstenedione, testosterone e diidrotestosterone).
Ciò può determinare nelle femmine la comparsa precoce di segni di iperandrogenismo (irsutismo, clitoride ingrossato, ambiguità dei genitali esterni nelle neonate e alopecia androgenetica in età adulta); ovaie, utero e salpingi sono nella norma, Nei maschi la patologia si manifesta clinicamente solo in un periodo successivo alla nascita attraverso un aumento patologico della velocità di crescita e pubertò precoce. Pertanto nei maschi la diagnosi della patologia alla nascita viene spesso misconosciuta a meno che non vi sia un concomitante deficit di aldosterone, che determina forti crisi ipotensive e perdita di sale.
Contemporaneamente, alla mancata sintesi del cortisolo si può aggiungere una insufficiente produzione di aldosterone che può determinare, nei casi più gravi, un concomitante squilibrio idroelettrolitico con ipovolemia e shock.
Inoltre, in questa patologia possono essere presenti anche altre alterazioni disfunzionali riguardanti non solo la corticale ma anche la midollare del surrene e ciò potrebbe spiegare la predisposizione di questi pazienti a sviluppare crisi surrenaliche acute in concomitanza di stati febbrili o eventi stressanti, anche se in terapia con una adeguata dose sostitutiva di glucocorticoidi.
Riguardo alla gravità della patologia si osserva un caleidoscopio di tipologie passando da un deficit più grave, detto deficit classico, che si manifesta già in epoca neonatale o nelle prime fasi dell’infanzia con virilizzazione e insufficienza surrenalica (con o senza perdita di sali), e uno meno grave, detto deficit non classico, che può essere asintomatico o associato solo a pochi segni di iperandrogenismo e che solitamente si manifesta più tardivamente (nella fase finale dell’infanzia o addirittura in età adulta). L’iperandrogenismo sia nella forma classica che in quella non classica porta allo sviluppo della PCOS, con conseguente oligomenorrea, amenorrea, soprattutto in adolescenza (16-32).
La gravità della malattia dipende dalla specifica mutazione di CYP21A2 e dal grado di deficit enzimatico.
Va ricordato, infine, che una attività enzimatica residua del solo 1-2% del normale può essere sufficiente, da sola, a modificare il fenotipo del paziente favorendo la forma clinica meno grave (virilizzante semplice) al posto di quella più grave (con perdita di sale).
SINTOMATOLOGIA: disidratazione, shock ipovolemico, virilizzazione
A) Circa il 75% dei pazienti con deficit classico di 21-idrossilasi è incapace di sintetizzare adeguate quantità di sia di cortisolo che di aldosterone con ipersecrezione surrenalica di DHEA, androstenedione e testosterone e conseguente alterazione dell’equilibrio elettrolitico e virilizzazione.
L’aldosterone è l’ormone che regola l’omeostasi del sodio e pertanto una sua mancanza determina, nei pazienti non trattati, una aumentata escrezione di sodio con conseguente iponatremia, ipovolemia e iperreninemia. Inoltre, siccome il potassio a livello renale viene scambiato con il sodio, l’elevata escrezione di quest’ultimo comporta un accumulo di potassio con conseguente iperkaliemia, soprattutto durante l’infanzia.
In questi pazienti il deficit di cortisolo complica ulteriormente il quadro clinico determinando un peggioramento della funzione cardiaca, una riduzione della risposta vascolare alle catecolammine ed una riduzione della filtrazione glomerulare.
Pertanto, la contemporanea assenza di cortisolo e aldosterone causa frequentemente disidratazione iponatriemica e, nei casi non trattati, anche shock ipovolemico. Clinicamente la perdita di sale si manifesta tipicamente fra il settimo e il quattordicesimo giorno di vita con vomito, perdita di peso, letargia, iponatremia e iperkaliemia.
I pazienti con il deficit classico di 21-idrossilasi con perdita di sale vengono identificati attraverso il dosaggio degli elettroliti, della renina e dell’aldosterone che mostrano, iperreninemia con bassi valori di aldosterone.
B) Forme con virilizzazione semplice: il paziente non è in grado di produrre sufficienti quantità di cortisolo ma è perfettamente in grado di produrre adeguate quantità di aldosterone e quindi di mantenere un corretto bilancio elettrolitico. Le femmine sono esposte, a partire dalla settima settimana di gestazione, ad elevate concentrazioni di androgeni. Questa sovraesposizione agli androgeni porta ad un grado variabile di virilizzazione del feto che, pertanto, alla nascita può avere delle ambiguità genitali tali da causare, addirittura, difficoltà nell’assegnazione del sesso fenotipico. Solitamente le ambiguità genitali più frequenti sono: clitoridomegalia, grandi labbra rugose e parzialmente fuse ed un seno urogenitale comune (invece di uretra e vagina separate). Al contrario, invece, i maschi affetti, alla nascita, non presentano, ovviamente, alcun segno genitale della patologia ad eccezione di una variabile, e comunque lieve, iperpigmentazione dello scroto ed un certo ingrandimento del pene. Pertanto nei maschi la diagnosi della patologia alla nascita viene spesso misconosciuta a meno che non vi sia un concomitante deficit di aldosterone, che determina forti crisi ipotensive e perdita di sale. Nei pazienti non trattati, o inadeguatamente trattati, l’esposizione per lungo tempo ad un eccesso di androgeni determina una rapida crescita corporea (effetto dovuto prevalentemente agli androgeni) ed un avanzamento dell’età ossea con una precoce chiusura delle cartilagini epifisarie (effetto dovuto prevalentemente agli estrogeni formatisi dalla aromatizzazione periferica degli androgeni) che si traduce infine con bassa statura patologica.
Anche il pubarca ed il telarca possono comparire precocemente.
Con il passare del tempo le dimensioni del clitoride possono continuare ad aumentare nelle femmine così come quelle del pene nel maschio, anche se il volume dei testicoli rimane sempre piccolo. L’iperandrogenismo, inoltre, sia nella forma classica che in quella non classica, può essere una condizione predisponente allo sviluppo della sindrome dell’ovaio policistico.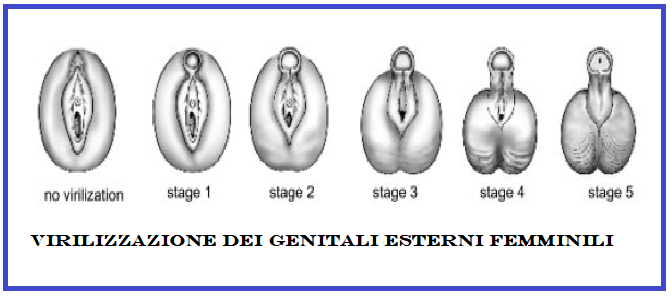
Nelle donne con qualsiasi forma di deficit di 21-idrossilasi possono comparire oligomenorrea o amenorrea, soprattutto durante il periodo adolescenziale. Inoltre, l’esposizione ad elevati livelli di androgeni durante il periodo prenatale può influenzare il successivo comportamento sessuale.
Inoltre, sebbene la fertilità possa essere notevolmente ridotta, nel caso in cui la produzione di androgeni surrenalici non sia adeguatamente soppressa con la terapia steroidea, molte donne con deficit di 21-idrossilasi sono ugualmente in grado di concepire e di portare a termine con successo una gravidanza. Si può pertanto affermare che circa l’80% delle donne con deficit virilizzante semplice ed approssimativamente il 60% di quelle con perdita di sale sono fertili.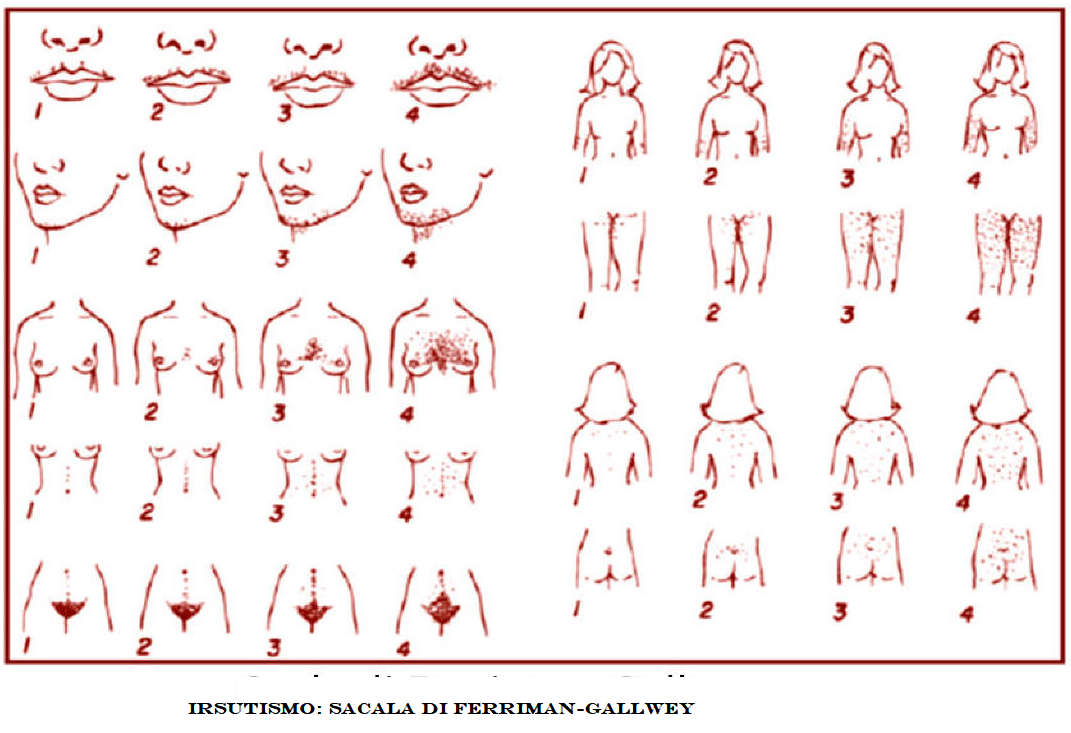
Gli uomini con deficit di 21-idrossilasi hanno molti meno problemi della funzione riproduttiva. Molti di essi, infatti, hanno uno spermiogramma nella norma ed una normale capacità riproduttiva. Tuttavia, nei maschi affetti una forma relativamente frequente di anormalità gonadica è lo sviluppo di tessuto surrenalico ectopico, il più delle volte a livello testicolare e quasi sempre bilaterale (testicular adrenal rest tissue). Embriologicamente parlando, sia le gonadi che i surreni originano dal mesoderma del tratto urogenitale e proprio per questo motivo il riscontro di tessuto surrenalico (adrenal remnants) è molto frequente nelle gonadi, soprattutto nel testicolo, sebbene possa riscontrarsi anche in altre parti del corpo (plesso celiaco, legamento largo e ovaie)
DIAGNOSTICA DI LABORATORIO
- cariotipo, in caso di ambiguità sessuale
- aldosteronemia
- reninemia
- 17-idrossi-progesterone basale: valori normali sono < 10 ng/ml; sono elevati nelle forme classiche di CAH
- Cortisolemia
- ACTH
- Na+
- K+
- Rx mano sinistra e polso: in caso di CAH si evidenzia un’età ossea più avanzata rispetto all’età anagrafica.
- USG, TAC, RMN del surrene
- Esame del gene della 21-idrossilasi mediante analisi sequenziale del DNA anche in epoca prenatale
- deficit di 21-idrossilasi: alta concentrazione sierica di 17-OH-progesterone (di solito >1000 ng/dL), bassa concentrazione di cortisolo e aldosterone, alta concentrazione di androgeni, di pregnantriolo urinario (metabolita del 17-OH-progesterone) e 17-chetosteroidi urinari.
- deficit 11-beta-idrossilasi: concentrazioni sieriche elevate di 11-desossicortisolo e desossicorticosterone e bassi livelli di cortisolo e corticosterone, elevati livelli di 17-chetosteroidi urinari/24 ore.
- deficit 3-beta-deidrogenasi: elevate concentrazioni di 17-OH-pregnenolone e DHEA che non vengono metabolizzati in 17-OH-progesterone e androstenedione rispettivamente.
- Villocentesi: diagnosi prenatale sul DNA estratto dai villi coriali (cariotipo e analisi diretta di CYP21, ottenibili intorno alla 11a-13a sett. di gestazione),
- Amniocentesi nel I° trimestre e dosaggio del 17-OH-progesterone nel liquido amniotico nel II° trimestre.
- Un test di screening neonatale (analisi del gene CYP21, il gene della 21-idrossilasi) è disponibile per la forma più comune di iperplasia surrenalica congenita; si effettua su sangue fetale ottenuto mediante cordocentesi o su sangue del neonato a 2-3 giorni dopo il parto.
- free fetal DNA in maternal plasma (36)
TERAPIA
Nel deficit classico di 21-idrossilasi la terapia prevede un trattamento a lungo termine con glucorticoidi per inibire l’eccessiva secrezione di CRH e ACTH da parte dell’ipotalamo e dell’ipofisi, rispettivamente.
Nei bambini il farmaco da preferire è l’idrocortisone (idrocortisone cpr 5 mg, 10 mg) somministrato ad un dosaggio di 10-20 mg per metro quadro di superficie corporea al giorno frazionata in tre dosi. Dosi superiori a 100 mg per metro quadro, invece, vanno somministrate durante gli episodi di insufficienza surrenalica acuta o comunque nelle situazioni a rischio di vita. Il monitoraggio della terapia viene effettuato attraverso il dosaggio dei livelli di 17-idrossi progesterone e di androstenedione. i livelli di 17-idrossi progesterone devono essere solo parzialmente soppressi a valori compresi fra 100 e 1000 ng/dl (3-30 nmol/l). La difficoltà maggiore nella terapia del deficit classico di 21-idrossilasi consiste nel trovare il giusto dosaggio farmacologico che impedisca l’iperandrogenismo senza causare, al tempo stesso, una condizione di ipercortisolismo; l’ipercortisolismo iatrogeno si rende clinicamente evidente per la comparsa di incremento ponderale, ridistribuzione tronculare dell’adipe, intolleranza glucidica, ipertensione e dislipidemia.
I bambini con il deficit classico di 21-idrossilasi con perdita di sale oltre alla terapia con glucocorticoidi richiedono un supplemento terapeutico con mineralcorticoidi (solitamente fludrocortisone acetato 0,1-0,2 mg/die) e cloruro di sodio (NaCl 1-2 g o 17-34 mmol/die). A differenza di quanto avviene per i glucocorticoidi il dosaggio terapeutico del fludrocortisone non è dipendente dal peso corporeo e pertanto non presenta sostanziali differenze fra il bambino e l’adulto. Per monitorare l’adeguatezza della terapia sostitutiva con mineralcorticoidi e cloruro di sodio bisogna valutare l’attività plasmatica reninica o direttamente la renina dosata con metodica RIA.
I pazienti con la forma virilizzante semplice non necessitano, per definizione, dei mineralcorticoidi ma, in alcuni casi, possono essere trattati con fludrocortisone perché in questo modo si raggiunge prima una maggior soppressione dell’asse surrenalico e di conseguenza si riduce il dosaggio di glucorticoidi richiesto per ottenere degli adeguati livelli di 17-idrossi progesterone.
Adolescenti e adulti: questi, infatti, devono essere trattati con prednisone (Deltacortene® cpr 5 mg, 25 mg) (ad un dosaggio variabile fra 5 e 7,5 mg/die frazionato in due dosi al giorno) o con desametazone (Decadron® cpr 0.5 mg, 0.75 mg) ad un dosaggio variabile fra 0,25 e 0,5 mg/die somministrato in una o due dosi al giorno. Questi pazienti devono essere strettamente monitorizzati per valutare la comparsa di segni di ipercortisolismo come strie rubre, osteopenia, ipertensione o rapido incremento ponderale.
Nel deficit non classico di 21-idrossilasi, invece, il trattamento farmacologico con glucocorticoidi non è indicato nei pazienti asintomatici, visto che gli effetti collaterali dei glucocorticoidi finirebbero per superarne i benefici.
I maschi con deficit non classico di 21-idrossilasi, solitamente, non richiedono alcun trattamento. Il trattamento con glicocorticoidi, invece, potrebbe essere indicato nei bambini maschi con pubertà precoce, con un aumento della velocità di crescita e con un avanzamento dell’età ossea, per ridurre il rischio di ipostaturismo. Invece nelle femmine, soprattutto nelle adolescenti e nelle giovani donne con segni di virilizzazione, può essere preso in considerazione anche un trattamento farmacologico alternativo, come l’utilizzo della pillola anticoncezionale estro-progestinca, di un farmaco con effetto antiandrogeno o di entrambi. La pillola anticoncezionale, invece, impedendo la sclerotizzazione e lo sviluppo di cisti ovariche, riduce, quindi, anche la sintesi degli androgeni. Utili le preparazioni farmacologiche che contengono un progestinico con proprietà antiandrogena tipo ciproterone (Diane® 21 cpr).
Lo spironolattone (Aldactone cpr 25 mg, 100 mg) e la flutamide (Eulexin® cpr 250 mg), il cui utilizzo non è formalmente approvato per questa patologia, sono, di fatto, gli antiandrogeni più utilizzati.
Terapia idratante, se necessaria
- Chirurgia plastica dei genitali, in caso di malformazioni e dopo aver effettuato il cariotipo. I lattanti femmine affetti possono richiedere una ricostruzione chirurgica con clitoroplastica riduttiva e vaginoplastica. Spesso, un ulteriore intervento chirurgico è necessario da adulti, ma con una cura e un’attenzione particolare per le problematiche psico-sessuali, è possibile ottenere una vita sessuale normale e il ripristino della fertilità.
TERAPIA IN GRAVIDANZA
La somministrazione di desametazone alla madre riduce le ambiguità genitali nei feti di sesso femminile affette da deficit di 21-idrossilasi impedendo così la virilizzazione del feto femmina.
Viene utilizzato il desametazone perché è un composto che non viene inattivato dall’enzima placentare 11β-idrossisteroido-deidrogenasi. Il dosaggio del farmaco è di 20 μg per Kg di peso corporeo al giorno (calcolato in base al peso corporeo della mamma prima dell’inizio della gravidanza) frazionato in tre dosi. Per prevenire la virilizzazione del feto è necessario somministrare il farmaco precocemente già dalla 7a settimana, epoca di inizio dello sviluppo dei genitali esterni, anche prima delle indagini sul feto. La terapia va proseguita se l’indagine genetica mostra che il feto è femmina e ha ereditato entrambe le copie del gene mutato, va sospesa se il feto è maschio, anche se affetto dalla patologia.
Va ricordato, inoltre, che il deficit di 21-idrossilasi è una patologia autosomica recessiva e pertanto, la probabilità che da due genitori portatori possa nascere una femmina affetta è solo di 1 su 8. Questo, chiaramente, vuol dire che ben 7 feti su 8 saranno sottoposti ad una terapia senza realmente averne bisogno.
Pertanto, per evitare la somministrazione di desametazone nei maschi o nelle femmine non affette è necessario effettuare una precoce ed accurata diagnosi genetica.
Alcune complicazioni potrebbero riguardare la madre; infatti lo sviluppo della sindrome di Cushing, l’incremento ponderale e lo sviluppo di ipertensione sono stati riscontrati nell’1% delle donne trattate in gravidanza. A ciò si aggiunge il fatto che l’efficacia della terapia prenatale nel prevenire la virilizzazione è ancora incerta. Dall’altro lato, tuttavia, la terapia con glucocorticoidi durante la gravidanza non sembra determinare rischi per il feto: il rischio di malformazioni, aborti e altre complicazioni è sovrapponibile a quella della popolazione generale.
References:
- Speiser PW1, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HF, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC; Endocrine Society. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Sep;95(9):4133-60. doi: 10.1210/jc.2009-2631
- Nieman LK. Uncommon causes of congenital adrenal hyperplasia. In: UpToDate, Basow, DS (Ed), UpToDate, Waltham, MA, 2013.
- Häggström, Mikael; Richfield, David (2014). “Diagram of the pathways of human steroidogenesis”. WikiJournal of Medicine. 1 (1). doi:10.15347/wjm/2014.005. ISSN 2002-4436.
- Bongiovanni, Alfred M.; Root, Allen W. (1963). “The Adrenogenital Syndrome”. The New England Journal of Medicine. 268 (23): 1283. doi:10.1056/NEJM196306062682308.
- White PC. Congenital adrenal hyperplasia and related disorders. In: Kliegman RM, Stanton BF, St. Geme JW III, Schor N, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 576.
- Yau M, Khattab A, Pina C, Yuen T, et al. Defects of andrenal steroidogenesis. In: Jameson JL, De Groot LJ, de Krester DM, et al, eds. Endocrinology: Adult and Pediatric. 7th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 104.
- C.J. Migeon, A.B. Wisniewski, Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency, Endocr. Metabol. Cl. North Am. 2001; 30, 1, 193-206
- P. Clayton, W.L. Miller et al., Consensus statement on 21-hydroxylase deficiency from the ESPE and the LWPES, Horm. Res. 2002; 58; 188-195
- Phenotype and genotype correlation of the microconversion from the CYP21A1P to the CYP21A2 gene in congenital adrenal hyperplasia. Torres, N., Mello, M.P., Germano, C.M., Elias, L.L., Moreira, A.C., Castro, M. Braz. J. Med. Biol. Res. (2003) [Pubmed]
- Preliminary investigation of mutations in 21-hydroxylase gene in patients with congenital adrenal hyperplasia in Russia. Evgrafov, O.V., Polyakov, A.V., Dzenis, I.G., Baharev, V.A. Hum. Mutat. (1995) [Pubmed]
- A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Tusie-Luna, M.T., Speiser, P.W., Dumic, M., New, M.I., White, P.C. Mol. Endocrinol. (1991)
- Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Perniola, R., Falorni, A., Clemente, M.G., Forini, F., Accogli, E., Lobreglio, G. Eur. J. Endocrinol. (2000)
- A de novo pathological point mutation at the 21-hydroxylase locus: implications for gene conversion in the human genome. Collier, S., Tassabehji, M., Sinnott, P., Strachan, T. Nat. Genet. (1993)
- Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. White, P.C., Speiser, P.W. Endocr. Rev. (2000)
- Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. Blanchong, C.A., Zhou, B., Rupert, K.L., Chung, E.K., Jones, K.N., Sotos, J.F., Zipf, W.B., Rennebohm, R.M., Yung Yu, C. J. Exp. Med. (2000) [Pubmed]
- Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Tusié-Luna, M.T., White, P.C. Proc. Natl. Acad. Sci. U.S.A. (1995)
- Single-nucleotide polymorphisms in intron 2 of CYP21P: evidence for a higher rate of mutation at CpG dinucleotides in the functional steroid 21-hydroxylase gene and application to segregation analysis in congenital adrenal hyperplasia. Jiddou, R.R., Wei, W.L., Sane, K.S., Killeen, A.A. Clin. Chem. (1999)
- Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB -TNXA hybrids in apparent large-scale gene conversions. Koppens, P.F., Hoogenboezem, T., Degenhart, H.J. Hum. Mol. Genet. (2002)
- Abundant adrenal-specific transcription of the human P450c21A “pseudogene”. Bristow, J., Gitelman, S.E., Tee, M.K., Staels, B., Miller, W.L. J. Biol. Chem. (1993)
- Diversity of the CYP21A2 gene: a 6.2-kb TaqI fragment and a 3.2-kb TaqI fragment mistaken as CYP21A1P. Lee, H.H., Tsai, F.J., Lee, Y.J., Yang, Y.C. Mol. Genet. Metab. (2006)
- Pitfalls of PCR-based genotyping in patients with 21-hydroxylase deficiency. Tsai, C.H., Lin, W.D., Tsai, F.J., Peng, C.T., Wu, J.Y. Acta paediatrica Taiwanica = Taiwan er ke yi xue hui za zhi. (2001) [Pubmed]
- The suppression effect of DNA sequences within the C4A region on the transcription activity of human CYP21. Chang, S.F., Cheng, C.L. Endocr. Res. (1998)
- A novel semiquantitative polymerase chain reaction/enzyme digestion-based method for detection of large scale deletions/conversions of the CYP21 gene and mutation screening in Turkish families with 21-hydroxylase deficiency. Tukel, T., Uyguner, O., Wei, J.Q., Yuksel-Apak, M., Saka, N., Song, D.X., Kayserili, H., Bas, F., Gunoz, H., Wilson, R.C., New, M.I., Wollnik, B. J. Clin. Endocrinol. Metab. (2003)
- Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. White, P.C., Vitek, A., Dupont, B., New, M.I. Proc. Natl. Acad. Sci. U.S.A. (1988)
- Pulsed field gel electrophoresis identifies a high degree of variability in the number of tandem 21-hydroxylase and complement C4 gene repeats in 21-hydroxylase deficiency haplotypes. Collier, S., Sinnott, P.J., Dyer, P.A., Price, D.A., Harris, R., Strachan, T. EMBO J. (1989)
- Two distinct areas of unequal crossingover within the steroid 21-hydroxylase genes produce absence of CYP21B. Donohoue, P.A., Jospe, N., Migeon, C.J., Van Dop, C. Genomics (1989)
- A novel frameshift mutation (141delT) in exon 1 of the 21-hydroxylase gene (CYP21) in a patient with the salt wasting form of congenital adrenal hyperplasia. Mutation in brief no. 255. Online. Krone, N., Braun, A., Roscher, A.A., Schwarz, H.P. Hum. Mutat. (1999)
- Sequences promoting the transcription of the human XA gene overlapping P450c21A correctly predict the presence of a novel, adrenal-specific, truncated form of tenascin-X. Tee, M.K., Thomson, A.A., Bristow, J., Miller, W.L. Genomics (1995)
- CYP21 pseudogene transcripts are much less abundant than those from the active gene in normal human adrenocortical cells under various conditions in culture. Endoh, A., Yang, L., Hornsby, P.J. Mol. Cell. Endocrinol. (1998)
- A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. Billerbeck, A.E., Bachega, T.A., Frazatto, E.T., Nishi, M.Y., Goldberg, A.C., Marin, M.L., Madureira, G., Monte, O., Arnhold, I.J., Mendonca, B.B. J. Clin. Endocrinol. Metab. (1999)
- R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Helmberg, A., Tusie-Luna, M.T., Tabarelli, M., Kofler, R., White, P.C. Mol. Endocrinol. (1992)
- CYP21 mutations and congenital adrenal hyperplasia. Lee, H. Clin. Genet. (2001)
- Identification of molecular defects causing congenital adrenal hyperplasia by cloning and differential hybridization of polymerase chain reaction-amplified 21-hydroxylase (CYP21) genes. Helmberg, A., Tabarelli, M., Fuchs, M.A., Keller, E., Dobler, G., Schnegg, I., Knorr, D., Albert, E., Kofler, R. DNA Cell Biol. (1992)
- Molecular analysis of CYP21 and C4 genes in Brazilian families with the classical form of steroid 21-hydroxylase deficiency. de-Araujo, M., Sanches, M.R., Suzuki, L.A., Guerra, G., Farah, S.B., de-Mello, M.P. Braz. J. Med. Biol. Res. (1996)
- How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. L’Allemand, D., Tardy, V., Grüters, A., Schnabel, D., Krude, H., Morel, Y. J. Clin. Endocrinol. Metab. (2000)
- New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, Khattab A, Liao GJ, Yau M, Kim SM, Chiu RW, Sun L, Zaidi M, Lo YM. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014 Jun;99(6):E1022-30. doi: 10.1210/jc.2014-1118.