
I surreni sono ghiandole posizionate al polo superiore di ciascun rene. Le ghiandole surrenali, utilizzando il colesterolo, sintetizzano glicocorticoidi (21 atomi di carbonio) tipo cortisolo, mineralcorticoidi (21 atomi di carbonio) tipo aldosterone e androgeni (19 atomi di carbonio) tipo DHEA, androstenedione, testosterone e diidrotestosterone.
Con il termine di iperplasia surrenalica congenita (CAH, Congenital Adrenal Hyperplasia) o sindrome adreno-genitale o s. da deficit di 21-idrossilasi si intende un gruppo di malattie autosomiche recessive (la malattia si manifesta soltanto se entrambe le copie del gene che esprime l’enzima sono alterate) che interessa 1/10.000 neonati.  E’ caratterizzata da disordini nella biosintesi degli ormoni steroidei, a causa di una mutazione in uno dei 5 enzimi (21-idrossilasi, 17-idrossilasi, 11-idrossilasi, 3-deidrogenasi, 20-22-desmolasi) coinvolti nel processo di sintesi. Questi enzimi sono prodotti dalla corteccia surrenalica, e nel 90% dei casi, la malattia dipende dal deficit dell’enzima 21-idrossilasi a sua volta causato da una mutazione nel gene CYP21A (detto anche CYP21P e P450c21A) che esprime la 21-idrossilasi. L’enzima 21-idrossilasi, appartiene alla famiglia dei citocromi P450, e converte il 17-OH-Progesterone in 11-desossicortisolo a sua volta trasformato in cortisolo ad opera della 11-ß-idrossilasi. In caso di deficit della 21-idrossilasi si ha dunque un accumulo di 17-OH-progesterone e deficit secretivo di cortisolo. L’ipocortisolemia induce, con meccanismo di feed-back positivo, un’ipersecrezione di ACTH da parte dell’ipofisi e quindi si instaura un circolo vizioso con iperplasia surrenalica, deficit di cortisolo e ulteriore accumulo di 17-OH-P, dal quale deriva iperproduzione di androgeni e conseguente sindrome adreno-genitale.
E’ caratterizzata da disordini nella biosintesi degli ormoni steroidei, a causa di una mutazione in uno dei 5 enzimi (21-idrossilasi, 17-idrossilasi, 11-idrossilasi, 3-deidrogenasi, 20-22-desmolasi) coinvolti nel processo di sintesi. Questi enzimi sono prodotti dalla corteccia surrenalica, e nel 90% dei casi, la malattia dipende dal deficit dell’enzima 21-idrossilasi a sua volta causato da una mutazione nel gene CYP21A (detto anche CYP21P e P450c21A) che esprime la 21-idrossilasi. L’enzima 21-idrossilasi, appartiene alla famiglia dei citocromi P450, e converte il 17-OH-Progesterone in 11-desossicortisolo a sua volta trasformato in cortisolo ad opera della 11-ß-idrossilasi. In caso di deficit della 21-idrossilasi si ha dunque un accumulo di 17-OH-progesterone e deficit secretivo di cortisolo. L’ipocortisolemia induce, con meccanismo di feed-back positivo, un’ipersecrezione di ACTH da parte dell’ipofisi e quindi si instaura un circolo vizioso con iperplasia surrenalica, deficit di cortisolo e ulteriore accumulo di 17-OH-P, dal quale deriva iperproduzione di androgeni e conseguente sindrome adreno-genitale.
Sintomatologia: I sintomi variano, a seconda del tipo di CAH e l’epoca di insorgenza.
I bambini con forme più lievi possono non avere segni o sintomi e la diagnosi in genere viene posta all’epoca di insorgenza della pubertà.
Nei neonati di sesso femminile con la forma più grave di CAH, l’esposizione in utero ad elevati livelli di androgeni possono causare anomalie genito-urinarie: le grandi labbra possono presentarsi saldate più o meno completamente sì da dar luogo a una formazione simile allo scroto; il clitoride si presenta nettamente ipertrofico sino ad assumere nei casi più gravi l’aspetto di un pene, mentre le vie urinarie e la vagina possono riunirsi in una sola cavità (seno urogenitale) con un unico sbocco all’esterno, spesso alla base del clitoride oppure fistoli uretero-vaginali. Altri sintomi frequentemente presenti sono:
- vomito
- diarrea
- Disidratazione
- perdita di peso
- shock
- aritmie cardiache
- ipogonadismo ipergonadotropo,
Le ragazze con la forma più lieve di CAH di solito hanno normali organi femminili riproduttivi (ovaie, utero e tube di Falloppio) e possono anche avere le seguenti modifiche:
- alterazioni mestruali o amenorrea
- comparsa precoce del pubarca
- irsutismo
- acne
- clitoridemegalia
Ragazzi con la forma più lieve appaiono spesso normali alla nascita. Tuttavia, essi spesso presentano: pubertà precoce e i sintomi possono includere:
- abbassamento della voce
- comparsa precoce dei peli pubici o ascellari
- pene ingrossato con testicoli normali
- muscoli ben sviluppati
In entrambi i sessi la statura si presenta normale o poco al di sotto della norma.
La diagnosi di CAH può anche essere posta in rapporto ai valori sierici o urinari di cortisolo, aldosterone, androgeni e precursori e metaboliti di tutti questi ormoni, come segue:
- deficit di 21-idrossilasi: alta concentrazione sierica di 17-OH-progesterone (di solito >1000 ng/dL), bassa concentrazione di mineralcorticoidi, alta concentrazione di androgeni, di pregnantriolo urinario (metabolita del 17-OH-progesterone) e 17-chetosteroidi urinari.
- deficit 11-beta-idrossilasi: concentrazioni sieriche elevate di 11-desossicortisolo e desossicorticosterone e bassi livelli di cortisolo e corticosterone, elevati livelli di 17-chetosteroidi urinari/24 ore.
- deficit 3-beta-deidrogenasi: elevate concentrazioni di 17-OH-prognenolone e DHEA che non vengono metabolizzati in 17-OH-progesterone e androstenedione rispettivamente.

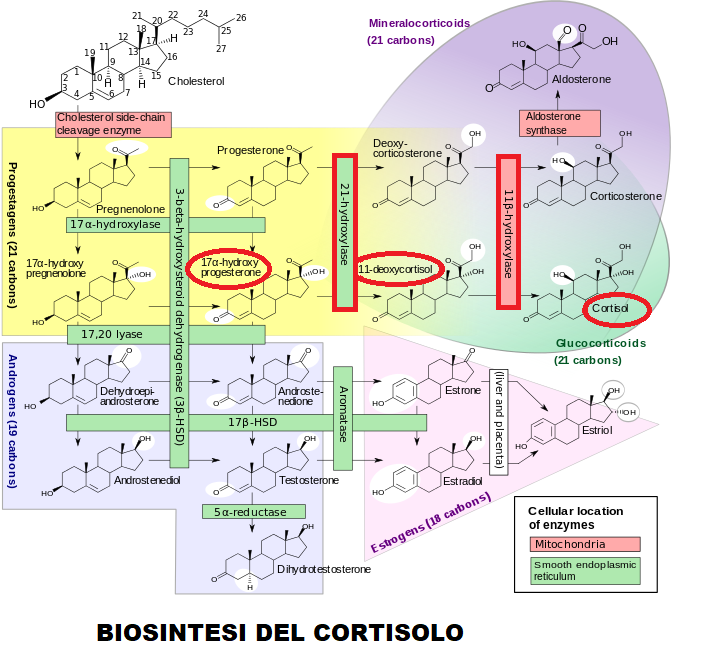
In base all’attività enzimatica di CYP-21, e alle caratteristiche fenotipiche delle persone affette, classifichiamo le forme di questa iperplasia in una forma classica e in una non classica
La cosiddetta forma classica si manifesta in epoca neonatale o nelle prime fasi dell’infanzia (in italia, 1:16000 nati). Può essere di 2 tipi:
- Con perdita di sale: In questo caso si ha deficit enzimatico totale, e non vengono prodotti ne cortisolo ne aldosterone (insufficienza surrenalica). Nei pazienti con deficit di cortisolo si ha un peggioramento della funzione cardiaca, con scarsa risposta vascolare alle catecolamine e una ridotta velocità di filtrazione glomerulare. L’assenza di entrambi gli ormoni si manifesta in maschi e femmine con vomito, diarrea, perdita di peso, disidratazione, shock.
- Virilizzazione semplice: In questi pazienti non si producono sufficienti quantità di cortisolo, ma si hanno adeguate quantità di aldosterone che consentono di avere un corretto bilancio elettrolitico. I disturbi sono dovuti principalmente all’eccesso di ormoni androgeni (iperandrogenismo). Nelle femmine, le manifestazioni cliniche sono quelle di un pseudoermafroditismo: come nella forma classica con perdita di sali, vi è sempre alla nascita un certo grado di virilizzazione dei genitali esterni. Successivamente si può rilevare la comparsa prematura di peluria pubica ed ascellare, di acne ed uno sviluppo delle masse muscolari e della tonalità della voce in senso maschile con una crescita accelerata.
La cosiddetta forma non classica può essere asintomatica o associata a pochi segni di iperandrogenismo, e in genere si manifesta tardivamente. Frequenze: (ebrei ashkenazi, 1:27; ispanici, 1:53; slavi 1:63; italiani, 1:333; caucasici, 1:1000). Questo suggerisce che più dell’1% della popolazione è eterozigote, e quindi portatrice dell’allele NC. La forma non classica può essere di 2 tipi:
- a insorgenza tardiva: In questi pazienti si verificano manifestazioni dovute a iperandrogenismo, come comparsa precoce di peluria pubica ed ascellare, modeste accelerazioni della crescita e della maturazione scheletrica, acne, irsutismo, PCOS. Nei maschi si può avere azospermia, oligospermia, infertilità.
- Criptica: Questa forma è completamente asintomatica, e prevalgono i segni clinici legati ad una condizione di iperandrogenismo moderato-lieve.
L’iperandrogenismo sia nella forma classica che in quella non classica porta allo sviluppo della policistosi ovarica, con conseguente oligomenorrea, amenorrea, soprattutto in adolescenza. Inoltre, sebbene la fertilità possa essere ridotta, anche se la produzione di androgeni non viene soppressa, una donna può concepire e portare a termine una gravidanza con successo. L’infertilità si ha solo nel 13% delle donne con deficit non classico di 21-idrossilasi,
Deficit congenito di 11-ß-idrossilasi: questo deficit è responsabile del 3-5% di tutti i casi di SAG. Il profilo steroideo è caratterizzato dal l’incremento dell’11-deossicortisolo (e dei 17-idrossicorticosteroidi urinari) e del deossicorticosterone. A causa dell’attività mineralcorticoide del deossicorticosterone, i pazienti presentano ritenzione salina e ipertensione arteriosa con alkalosi ipokaliemica. L’attività reninica plasmatica è bassa. Si realizza anche virilizzazione. Il trattamento consiste nella terapia sostitutiva con cortisolo; può anche essere necessaria quella con mineralcorticoidi.
Deficit congenito di 3-b-OH-steroido-deidrogenasi: questo disturbo molto raro comporta l’accumulo di DHEA, che viene convertito in testosterone perifericamente nel tessuto extrasurrenalico. Il trattamento consiste anche qui di glucocorticoidi con mineralcorticoidi quando necessario.
Deficit congenito di colesterolo-desmolasi e di 17-a-idrossilasi: questi disturbi provocano la virilizzazione dei lattanti femmine affetti e l’ipovirilizzazione di quelli maschi.
Deficit congenito di corticosterone 18-metilossidasi di tipo II: la manifestazione è quella tipica della carenza di aldosterone: iperkaliemia cronica e bassa aldosteronemia. Non sono presenti alterazioni della differenziazione sessuale.
Diagnostica di laboratorio:
- cariotipo, in caso di ambiguità sessuale
- aldosteronemia
- reninemia
- 17-idrossi-progesterone basale: valori normali sono < 10 ng/ml; sono elevati nelle forme classiche di CAH
- Cortisolemia
- ACTH elevato
- iponatriemia
- iperkaliemia
- Rx mano sinistra e polso: in caso di CAH si evidenzia un’età ossea più avanzata rispetto all’età anagrafica.
- USG, TAC, RMN del surrene
- Esame del gene della 21-idrossilasi mediante analisi sequenziale del DNA anche in epoca prenatale su cellule fetali prelevate mediante amniocentesi o villocentesi. La ricerca di anomalie geniche può aiutare a confermare la diagnosi in casi dubbi, ma raramente è indispensabile.
Possibili Complicazioni:
- Ipertensione arteriosa ipercalemica
- Ipoglicemia
- Iposodiemia
- Iperpotassiemia
Terapia: L’obiettivo del trattamento è quello di restituire i livelli ormonali alla normalità, per quanto possibile.
- Chirurgia plastica dei genitali, in caso di malformazioni e dopo aver effettuato il cariotipo. I lattanti femmine affetti possono richiedere una ricostruzione chirurgica con clitoroplastica riduttiva e vaginoplastica. Spesso, un ulteriore intervento chirurgico è necessario da adulti, ma con una cura e un’attenzione particolare per le problematiche psico-sessuali, è possibile ottenere una vita sessuale normale e il ripristino della fertilità.
- Terapia ormonale sostitutiva per tutta la vita , con farmaci a base di cortisonici e mineralcorticoidi (idrocortisone, cortisone acetato o prednisone). La somministrazione orale di idrocortisone (da 15 a 25 mg/m2 /die in 3 dosi) o prednisone (3-4 mg/m2 /die in 2 dosi) viene adeguata per mantenere il livello sierico dei precursori androgeni nel range appropriato per l’età. della paziente. Il cortisone acetato IM 18-36 mg/m2 ogni 3 giorni può anche essere utilizzato nei lattanti quando la terapia orale non è affidabile. La terapia è finalizzata alla normalizzazione sia dell’androstendione, del 17-OH-progesterone e dell’attività reninica, dei metaboliti urinari (17-ketosteroidi e pregnantriolo). Il fluidrocortisone per via orale (0,1 mg/die) va somministrato se c’è perdita di sale. I lattanti spesso richiedono un supplemento di sale per os. È difficile il monitoraggio stretto durante la terapia. L’ipertrattamento con glucocorticoidi determina la malattia di Cushing iatrogena, che si manifesta nell’infanzia con obesità, crescita ridotta e ritardo dell’età ossea. L’ipo-trattamento con glucocorticoidi non riesce a sopprimere la secrezione di ACTH con conseguente iperandrogenismo, che si manifesta nell’infanzia con virilizzazione e velocità di crescita superiore alla norma e infine interruzione precoce dell’accrescimento con bassa statura finale. Bisogna assicurare la compliance al trattamento, l’accrescimento deve essere rigorosamente monitorizzato e l’età ossea valutata ogni anno.
- Terapia idratante, se necessaria.
Prevenzione: un’anamnesi familiare positiva per iperplasia surrenalica congenita (di qualsiasi tipo), dovrebbe obbligare a prendere in considerazione la consulenza genetica prenatale con villocentesi o amniocentesi nel I° trimestre e dosaggio del 17-OH-progesterone nel liquido amniotico nel II° trimestre. Un test di screening neonatale (analisi del gene CYP21, il gene della 21-idrossilasi) è disponibile per la forma più comune di iperplasia surrenalica congenita; si effettua su sangue fetale ottenuto mediante cordocentesi o su sangue del neonato a 2-3 giorni dopo il parto.
References:
- Speiser PW1, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HF, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC; Endocrine Society. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010 Sep;95(9):4133-60. doi: 10.1210/jc.2009-2631
- Nieman LK. Uncommon causes of congenital adrenal hyperplasia. In: UpToDate, Basow, DS (Ed), UpToDate, Waltham, MA, 2013.
- Häggström, Mikael; Richfield, David (2014). “Diagram of the pathways of human steroidogenesis”. WikiJournal of Medicine. 1 (1). doi:10.15347/wjm/2014.005. ISSN 2002-4436.
- Bongiovanni, Alfred M.; Root, Allen W. (1963). “The Adrenogenital Syndrome”. The New England Journal of Medicine. 268 (23): 1283. doi:10.1056/NEJM196306062682308.
- White PC. Congenital adrenal hyperplasia and related disorders. In: Kliegman RM, Stanton BF, St. Geme JW III, Schor N, eds. Nelson Textbook of Pediatrics. 20th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 576.
- Yau M, Khattab A, Pina C, Yuen T, et al. Defects of andrenal steroidogenesis. In: Jameson JL, De Groot LJ, de Krester DM, et al, eds. Endocrinology: Adult and Pediatric. 7th ed. Philadelphia, PA: Elsevier Saunders; 2016:chap 104.
- C.J. Migeon, A.B. Wisniewski, Congenital adrenal hyperplasia owing to 21-hydroxylase deficiency, Endocr. Metabol. Cl. North Am. 2001; 30, 1, 193-206
- P. Clayton, W.L. Miller et al., Consensus statement on 21-hydroxylase deficiency from the ESPE and the LWPES, Horm. Res. 2002; 58; 188-195
- Phenotype and genotype correlation of the microconversion from the CYP21A1P to the CYP21A2 gene in congenital adrenal hyperplasia. Torres, N., Mello, M.P., Germano, C.M., Elias, L.L., Moreira, A.C., Castro, M. Braz. J. Med. Biol. Res. (2003) [Pubmed]
- Preliminary investigation of mutations in 21-hydroxylase gene in patients with congenital adrenal hyperplasia in Russia. Evgrafov, O.V., Polyakov, A.V., Dzenis, I.G., Baharev, V.A. Hum. Mutat. (1995) [Pubmed]
- A mutation (Pro-30 to Leu) in CYP21 represents a potential nonclassic steroid 21-hydroxylase deficiency allele. Tusie-Luna, M.T., Speiser, P.W., Dumic, M., New, M.I., White, P.C. Mol. Endocrinol. (1991)
- Organ-specific and non-organ-specific autoantibodies in children and young adults with autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED). Perniola, R., Falorni, A., Clemente, M.G., Forini, F., Accogli, E., Lobreglio, G. Eur. J. Endocrinol. (2000)
- A de novo pathological point mutation at the 21-hydroxylase locus: implications for gene conversion in the human genome. Collier, S., Tassabehji, M., Sinnott, P., Strachan, T. Nat. Genet. (1993)
- Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. White, P.C., Speiser, P.W. Endocr. Rev. (2000)
- Deficiencies of human complement component C4A and C4B and heterozygosity in length variants of RP-C4-CYP21-TNX (RCCX) modules in caucasians. The load of RCCX genetic diversity on major histocompatibility complex-associated disease. Blanchong, C.A., Zhou, B., Rupert, K.L., Chung, E.K., Jones, K.N., Sotos, J.F., Zipf, W.B., Rennebohm, R.M., Yung Yu, C. J. Exp. Med. (2000) [Pubmed]
- Gene conversions and unequal crossovers between CYP21 (steroid 21-hydroxylase gene) and CYP21P involve different mechanisms. Tusié-Luna, M.T., White, P.C. Proc. Natl. Acad. Sci. U.S.A. (1995)
- Single-nucleotide polymorphisms in intron 2 of CYP21P: evidence for a higher rate of mutation at CpG dinucleotides in the functional steroid 21-hydroxylase gene and application to segregation analysis in congenital adrenal hyperplasia. Jiddou, R.R., Wei, W.L., Sane, K.S., Killeen, A.A. Clin. Chem. (1999)
- Carriership of a defective tenascin-X gene in steroid 21-hydroxylase deficiency patients: TNXB -TNXA hybrids in apparent large-scale gene conversions. Koppens, P.F., Hoogenboezem, T., Degenhart, H.J. Hum. Mol. Genet. (2002)
- Abundant adrenal-specific transcription of the human P450c21A “pseudogene”. Bristow, J., Gitelman, S.E., Tee, M.K., Staels, B., Miller, W.L. J. Biol. Chem. (1993)
- Diversity of the CYP21A2 gene: a 6.2-kb TaqI fragment and a 3.2-kb TaqI fragment mistaken as CYP21A1P. Lee, H.H., Tsai, F.J., Lee, Y.J., Yang, Y.C. Mol. Genet. Metab. (2006)
- Pitfalls of PCR-based genotyping in patients with 21-hydroxylase deficiency. Tsai, C.H., Lin, W.D., Tsai, F.J., Peng, C.T., Wu, J.Y. Acta paediatrica Taiwanica = Taiwan er ke yi xue hui za zhi. (2001) [Pubmed]
- The suppression effect of DNA sequences within the C4A region on the transcription activity of human CYP21. Chang, S.F., Cheng, C.L. Endocr. Res. (1998)
- A novel semiquantitative polymerase chain reaction/enzyme digestion-based method for detection of large scale deletions/conversions of the CYP21 gene and mutation screening in Turkish families with 21-hydroxylase deficiency. Tukel, T., Uyguner, O., Wei, J.Q., Yuksel-Apak, M., Saka, N., Song, D.X., Kayserili, H., Bas, F., Gunoz, H., Wilson, R.C., New, M.I., Wollnik, B. J. Clin. Endocrinol. Metab. (2003)
- Characterization of frequent deletions causing steroid 21-hydroxylase deficiency. White, P.C., Vitek, A., Dupont, B., New, M.I. Proc. Natl. Acad. Sci. U.S.A. (1988)
- Pulsed field gel electrophoresis identifies a high degree of variability in the number of tandem 21-hydroxylase and complement C4 gene repeats in 21-hydroxylase deficiency haplotypes. Collier, S., Sinnott, P.J., Dyer, P.A., Price, D.A., Harris, R., Strachan, T. EMBO J. (1989)
- Two distinct areas of unequal crossingover within the steroid 21-hydroxylase genes produce absence of CYP21B. Donohoue, P.A., Jospe, N., Migeon, C.J., Van Dop, C. Genomics (1989)
- A novel frameshift mutation (141delT) in exon 1 of the 21-hydroxylase gene (CYP21) in a patient with the salt wasting form of congenital adrenal hyperplasia. Mutation in brief no. 255. Online. Krone, N., Braun, A., Roscher, A.A., Schwarz, H.P. Hum. Mutat. (1999)
- Sequences promoting the transcription of the human XA gene overlapping P450c21A correctly predict the presence of a novel, adrenal-specific, truncated form of tenascin-X. Tee, M.K., Thomson, A.A., Bristow, J., Miller, W.L. Genomics (1995)
- CYP21 pseudogene transcripts are much less abundant than those from the active gene in normal human adrenocortical cells under various conditions in culture. Endoh, A., Yang, L., Hornsby, P.J. Mol. Cell. Endocrinol. (1998)
- A novel missense mutation, GLY424SER, in Brazilian patients with 21-hydroxylase deficiency. Billerbeck, A.E., Bachega, T.A., Frazatto, E.T., Nishi, M.Y., Goldberg, A.C., Marin, M.L., Madureira, G., Monte, O., Arnhold, I.J., Mendonca, B.B. J. Clin. Endocrinol. Metab. (1999)
- R339H and P453S: CYP21 mutations associated with nonclassic steroid 21-hydroxylase deficiency that are not apparent gene conversions. Helmberg, A., Tusie-Luna, M.T., Tabarelli, M., Kofler, R., White, P.C. Mol. Endocrinol. (1992)
- CYP21 mutations and congenital adrenal hyperplasia. Lee, H. Clin. Genet. (2001)
- Identification of molecular defects causing congenital adrenal hyperplasia by cloning and differential hybridization of polymerase chain reaction-amplified 21-hydroxylase (CYP21) genes. Helmberg, A., Tabarelli, M., Fuchs, M.A., Keller, E., Dobler, G., Schnegg, I., Knorr, D., Albert, E., Kofler, R. DNA Cell Biol. (1992)
- Molecular analysis of CYP21 and C4 genes in Brazilian families with the classical form of steroid 21-hydroxylase deficiency. de-Araujo, M., Sanches, M.R., Suzuki, L.A., Guerra, G., Farah, S.B., de-Mello, M.P. Braz. J. Med. Biol. Res. (1996)
- How a patient homozygous for a 30-kb deletion of the C4-CYP 21 genomic region can have a nonclassic form of 21-hydroxylase deficiency. L’Allemand, D., Tardy, V., Grüters, A., Schnabel, D., Krude, H., Morel, Y. J. Clin. Endocrinol. Metab. (2000)
- New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, Khattab A, Liao GJ, Yau M, Kim SM, Chiu RW, Sun L, Zaidi M, Lo YM. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014 Jun;99(6):E1022-30. doi: 10.1210/jc.2014-1118.