
Ultimo aggiornamento 2022-03-13 20:41:37
Si definisce amenorrea primaria l’assenza del menarca oltre i 16-18 anni oppure 2 anni dopo lo sviluppo puberale (telarca, pubarca e crescita ossea e staturale) che avviene all’età di circa 12-13 anni nell’Europa Occidentale.
Criptomenorrea invece è il termine utilizzato per definire una mestruazione regolarmente costituita in cui non si verifica la fuoriuscita delle perdite ematiche a causa di ostacoli meccanici come imene imperforato, setti vaginali completi, agenesia vaginale, stenosi o assenza del collo dell’utero, sinechie uterine (1).
INDICE:
- Difetti anatomici utero-vaginali
- Alterata produzione congenita di Gn-RH
- Disgenesie gonadiche
- Sindrome della sella vuota
- Sindrome di DiGeorge
DIFETTI ANATOMICI UTERO-VAGINALI
Atresia del canale cervico-istmico: amenorrea primaria con ematometra senza ematocolpo.

ematometra
L’esame ginecologico evidenzia una vagina normale che termina in un “cul de sac” in cui la cervice uterina è assente. si palpa l’utero di volume aumentato e consistenza diminuita, quasi cistica. L’same US evidenzia un utero con isterometria molto aumentata a causa di ematometra che distende la cavità uterina. La terapia è chirurgica: tracheloplastica dove possibile.
La S. di Rokintasky-Kuster-Hauser è una condizione di agenesia utero-vaginale dovuta a mancata fusione o mancata canalizzazione dei dotti di Mϋller durante l’organogenesi. Il cariotipo è normale e l’etiologia è da ricondurre all’esposizione del feto in utero a fattori tosso-infettivi (17-25).
L’utero è rappresentato da corni uterini atrofici e i ⅔ superiori della vagina da tessuto fibroso; non vi è imene. Le ovaie sono normali e normofunzionanti. Presenti pubarca e telarca, non vi sono ritardi di crescita o disturbi mentali ma frequentemente si possono riscontrare malformazioni renali e vertebrali fino a configurare la “Associazione MURCS”, una condizione molto rara caratterizzata dalla contemporanea presenza di diverse anomalie di sviluppo.
La terapia è di tipo chirurgico: creazione di neo-vagina artificiale utilizzando lembi cutanei (metodo McIndoe) (3) o intestinali o mediante la tecnica di Vecchietti (4,5). La tecnica di Vecchietti prevede l’inserimento, a livello del fondo cieco della vagina, di una dispositivo di forma ovalare che viene successivamente messo in trazione con fili che dall’interno dell’addome fuoriescono all’esterno e sono progressivamente stirati in alto di 1 cm al giorno mediante un semplice apparecchio meccanico. La gravidanza non è possibile per queste donne che possono, nei paesi dove ciò è permesso, usufruire della fecondazione in vitro con propri ovociti e della maternità surrogata (“utero in affitto”).
Trapianto di utero: Il trapianto di utero è stato effettuato per la prima volta nel 2000, in Arabia Saudita, su una donna di 26 anni prelevando l’organo da donatrice deceduta. Per sopravvenute complicanze trombotiche, l’utero è stato successivamente rimosso. In Svezia, presso l‘università di Goteborg sono stati effettuati con successo 2 trapianti di utero da madre a figlia, di quest’ultime ultime una era affetta da RKHS e l’altra era stata precedentemente sottoposta ad isterectomia per K cervicale. In Italia questo tipo di intervento è proibito. Il trapianto di utero da donatrice vivente richiederebbe in Italia un’apposita legge come per i trapianti di polmone e intestino. L’intervento sperimentale sui topi ha riscontrato una percentuale di successo in termini di gravidanze del 10%.
Vagina biotech: Per la prima volta al mondo, nel 2006, Il tessuto mucoso è stato ricostruito in provetta utilizzando cellule autologhe delle stesse pazienti ed è stato poi trapiantato in 2 pazienti. L’intervento di ricostruzione biotech e il successivo trapianto sono stati effettuati con successo da Cinzia Marchese del dipartimento di medicina sperimentale dell’università La Sapienza di Roma e da Pierluigi Benedetti Panici, Direttore del DAI di Ginecologia e Ostetricia dello stesso ateneo.
Malformazioni vulvo-vaginali:
– imene imperforato: determina una falsa amenorrea (criptomenorrea).  Clinicamente la giovane lamenta dolori pelvici con riacutizzazioni cicliche e presenta un ematocolpo, l’imene è teso, bombato. Nelle neonate e nelle bambine pre-puberali si può presentare una situazione simile per la raccolta delle secrezioni vaginali (mucocolpo). In queste bambine possono verificarsi difficoltà di svuotamento della vescica e conseguenti infezioni vescicali ed ureterali ricorrenti. Una incisione imeneale risolve il problema (15,16).
Clinicamente la giovane lamenta dolori pelvici con riacutizzazioni cicliche e presenta un ematocolpo, l’imene è teso, bombato. Nelle neonate e nelle bambine pre-puberali si può presentare una situazione simile per la raccolta delle secrezioni vaginali (mucocolpo). In queste bambine possono verificarsi difficoltà di svuotamento della vescica e conseguenti infezioni vescicali ed ureterali ricorrenti. Una incisione imeneale risolve il problema (15,16).
Diaframma vaginale completo: stessa sintomatologia e trattamento.
Aplasie vaginali: quasi sempre associata ad assenza dell’utero e tube rudimentali (S. di Rokitansky). Non c’è la ritenzione mestruale ma la funzionalità ovarica è normale. Trattamento come per la RKHS.
Aplasia vaginale con presenza di utero funzionale: è eccezionale da sola. Terapia: neovagina; eventuale amputazione del collo se l’utero è atresico.
Sinechie delle piccole labbra: in seguito a flogosi vulvare non curata, le piccole labbra possono collabire strettamente ed impedire il deflusso delle secrezioni vaginali e/ o del sangue mestruale. La terapia è esclusivamente medica e prevede l’applicazione locale, mattina e sera tutti i giorni per 2 mesi, di creme vaginali a base di estrogeni a concentrazioni crescenti: Colpotrophine® (promestriene 1 g/100 gr), Colpogyn® (estriolo 12.5 mg/100 gr) e quindi Premarin® (estrogeni coniugati 62, 5 mg/100 gr). La vaselina applicata per ulteriori 30 giorni servirà ad evitare le recidive. Altri AA. consigliano di utilizzare solo creme idratanti (Alkagin® gel vaginale, Echigin® gel vaginale) per evitare gli effetti collaterali della terapia con estrogeni. Se necessario si applicheranno anche pomate specifiche per flogosi (Ledercort A pomata®, Altosone pomata®, Ecoval 70 lozione®), infezioni batteriche (Gentalyn beta crema mite®) e micotiche (Micotef®, Talsutin®, AB® crema vaginale, violetto di genziana in soluzione acquosa all’1%).
***************************************************************************
Amenorrea primaria da alterata produzione congenita di Gn-RH:
- Sindrome di Kallmann
- Sindrome di Prader-Willi
- Sindrome di Laurence-Moon-Biedl
- Ipogonadismo ipogonadotropo isolato
- Resistenza recettoriale al Gn-RH
- Mutazione DAX-1
*********************************************************************
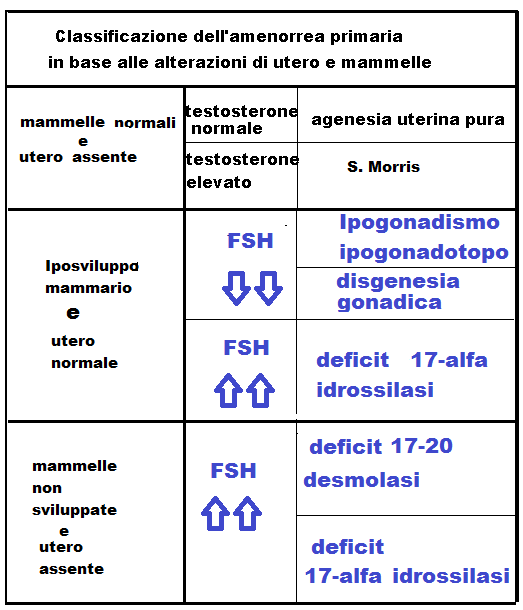
DISGENESIE GONADICHE:
1 – Sindrome di Turner: (45,X0) e Sindromi turneriane
2 – S. di Morris o femminilizzazione testicolare o S. da deficit recettoriale agli androgeni (45,XY)
3 – Sindrome di Swyer (46,XY)
—————————————————————–
Sindrome della sella vuota (empty sella sindrome): è un’affezione congenita o secondaria a traumi o radiazioni, in cui la sella turcica non è occupata dall’ipofisi ma da una estroflessione della cisterna soprachiasmatica.
Sindrome di DiGeorge o microdelezione del cromosoma 21q11.2: è una delle poche malattie da carenza immunitaria da deficit da  linfociti T descritta per la prima volta nel 1960 dall’endocrinologo Angelo DiGeorge. I sintomi si manifestano subito dopo la nascita, determinata da un difetto embrio-morfogenetico. La patogenesi di questa rara patologia è da riferire a delezione del cromosoma 21q11.2 o mutazione del gene TBX1. I pazienti presentano ipoplasia o aplasia del timo e delle paratiroidi, con la caratteristica sintomatologia ipocalcemica (tetania, convulsioni) che si verifica nelle prime 24 ore di vita, malformazioni cardiache con difetti settali e interruzione dell’arco aortico e mancata chiusura del dotto arterioso, basso impianto auricolare, ipoplasia mandibolare, palatoschisi e labbro leporino. L’ipocalcemia di questi soggetti è permanente. Questa sindrome causa l’insorgenza di infezioni spesso letali. In un piccolo numero di casi è indicato il trapianto di timo fetale il più presto possibile e l’ipoparatiroidismo va curato con calcio e vitamina D o paratormone. Nel 30% dei casi sono presenti anomalie del sistema genito-urinario con amenorrea primaria o, più raramente, secondaria (12-14).
linfociti T descritta per la prima volta nel 1960 dall’endocrinologo Angelo DiGeorge. I sintomi si manifestano subito dopo la nascita, determinata da un difetto embrio-morfogenetico. La patogenesi di questa rara patologia è da riferire a delezione del cromosoma 21q11.2 o mutazione del gene TBX1. I pazienti presentano ipoplasia o aplasia del timo e delle paratiroidi, con la caratteristica sintomatologia ipocalcemica (tetania, convulsioni) che si verifica nelle prime 24 ore di vita, malformazioni cardiache con difetti settali e interruzione dell’arco aortico e mancata chiusura del dotto arterioso, basso impianto auricolare, ipoplasia mandibolare, palatoschisi e labbro leporino. L’ipocalcemia di questi soggetti è permanente. Questa sindrome causa l’insorgenza di infezioni spesso letali. In un piccolo numero di casi è indicato il trapianto di timo fetale il più presto possibile e l’ipoparatiroidismo va curato con calcio e vitamina D o paratormone. Nel 30% dei casi sono presenti anomalie del sistema genito-urinario con amenorrea primaria o, più raramente, secondaria (12-14).
References:
- Dewhurts: Trattato di Ostetricia e Ginecologia EMSI editore, 2012.
- Massouras HG, Coutifaris B, Kalogirou D.: “Management of uterine adhesions with ‘Massouras Duck’s Foot’ and ‘Butterfly’ IUDs”. Contracept Deliv Syst.1982 Jan;3(1):25-38.
- S. Saraf; P. Saraf (2007). McIndoe Vaginoplasty: Revisited. The Internet Journal of Gynecology and Obstetrics 6(2).
- Vecchietti G (1965). [Creation of an artificial vagina in Rokitansky-Küster-Hauser syndrome]. Attual Ostet Ginecol 11 (2): 131–47 (in Italian).
- Fedele L, Bianchi S, Tozzi L, Borruto F, Vignali M (1996). A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil. Steril. 66 (5): 854–7
- K. Morcel, D. Guerrier, T. Watrin, I. Pellerin, and J. Levêque, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: clinical description and genetics,” Journal de Gynecologie Obstetrique et Biologie de la Reproduction, vol. 37, no. 6, pp. 539–546, 2008.
- J. E. Griffin, C. Edwards, and J. D. Madden, “Congenital absence of the vagina. The Mayer-Rokitansky-Küster-Hauser syndrome,” Annals of Internal Medicine, vol. 85, no. 2, pp. 224–236, 1976.
- K. Fisher, R. H. Esham, and I. Thorneycroft, “Scoliosis associated with typical Mayer-Rokitansky-Küster-Hauser syndrome,” Southern Medical Journal, vol. 93, no. 2, pp. 243–246, 2000.
- A. Pizzo, A. Fattori, C. Dugo, M. T. Mastroeni, C. Moscheo, and N. Dugo, “Syndrome of Rokitansky-Kunster-Hauser-Mayer: a description of four cases,” Minerva Ginecologica, vol. 59, no. 1, p. 95, 2007. View at Scopus
- E. H. Strübbe, C. W. Cremers, W. N. Willemsen, R. Rolland, and C. J. Thijn, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: two separate entities?” Clinical Dysmorphology, vol. 3, no. 3, pp. 192–199, 1994.
- E. H. Strübbe, J. A. Lemmens, C. J. Thijn, W. N. Willemsen, and B. S. van Toors, “Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome,” Skeletal Radiology, vol. 21, no. 7, pp. 459–462, 1992.
- Caridad de las Mercedes Tablada Borrero. Síndrome de Di George (Aplasia o Hipoplasia tímica). Multimed 2011; 15(1).
- Fomin A, Pastorino AC, Kim Chong A, Pereira CA, Carneiro-Sampaio M, Abe-Jacob CM. DiGeorge Syndrome: a not so rare disease. Clinics [artículo en Internet]. 2010 [Consultado 2 febrero 2011]; 65(9): [865-869]. Disponible en: http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1807
- Di George AM. Congenital absence of the thymus and its immunological consequences: concurrence with congenital hypoparathyroidism. Birth Defects 1968; 4: 116-21.
- S. Jean. “Physical Examination of the Child and Adolescent” (2000) in Evaluation of the Sexually Abused Child: A Medical Textbook and Photographic Atlas, Second edition, Oxford University Press. 62
- Sally E. Perlman, Nakajima, Steven T. and Hertweck, S. Paige, Clinical protocols in pediatric and adolescent gynecology, Parthenon, 2004, p. 131.
- S. Saraf; P. Saraf (2007). McIndoe Vaginoplasty: Revisited. The Internet Journal of Gynecology and Obstetrics 6(2).
- Vecchietti G (1965). [Creation of an artificial vagina in Rokitansky-Küster-Hauser syndrome]. Attual Ostet Ginecol 11 (2): 131–47 (in Italian). PMID 5319813.
- ^ Fedele L, Bianchi S, Tozzi L, Borruto F, Vignali M (1996). A new laparoscopic procedure for creation of a neovagina in Mayer-Rokitansky-Kuster-Hauser syndrome. Fertil. Steril. 66 (5): 854–7
- K. Morcel, D. Guerrier, T. Watrin, I. Pellerin, and J. Levêque, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome: clinical description and genetics,” Journal de Gynecologie Obstetrique et Biologie de la Reproduction, vol. 37, no. 6, pp. 539–546, 2008.
- J. E. Griffin, C. Edwards, and J. D. Madden, “Congenital absence of the vagina. The Mayer-Rokitansky-Küster-Hauser syndrome,” Annals of Internal Medicine, vol. 85, no. 2, pp. 224–236, 1976.
- K. Fisher, R. H. Esham, and I. Thorneycroft, “Scoliosis associated with typical Mayer-Rokitansky-Küster-Hauser syndrome,” Southern Medical Journal, vol. 93, no. 2, pp. 243–246, 2000.
- A. Pizzo, A. Fattori, C. Dugo, M. T. Mastroeni, C. Moscheo, and N. Dugo, “Syndrome of Rokitansky-Kunster-Hauser-Mayer: a description of four cases,” Minerva Ginecologica, vol. 59, no. 1, p. 95, 2007.
- E. H. Strübbe, C. W. Cremers, W. N. Willemsen, R. Rolland, and C. J. Thijn, “The Mayer-Rokitansky-Küster-Hauser (MRKH) syndrome without and with associated features: two separate entities?” Clinical Dysmorphology, vol. 3, no. 3, pp. 192–199, 1994.
- E. H. Strübbe, J. A. Lemmens, C. J. Thijn, W. N. Willemsen, and B. S. van Toors, “Spinal abnormalities and the atypical form of the Mayer-Rokitansky-Küster-Hauser syndrome,” Skeletal Radiology, vol. 21, no. 7, pp. 459–462, 1992.