Ultimo aggiornamento 13/11/2023
EPIDEMIOLOGIA – Il cancro dell’ovaio interessa l’1.7% di tutta la popolazione femminile mondiale con 165.000 nuovi casi ogni anno nel mondo, di cui 4.000 in Italia. E’ la seconda per frequenza tra le neoplasie ginecologiche ed è quella con la più alta mortalità (70%) a causa soprattutto della diagnosi tardiva (1-5).
I fattori etiologici del cancro ovarico sono ancora da definire completamente anche se sono state individuate specifiche alterazioni genetiche, endocrine e fattori familiari.
Nonostante la bassa incidenza, la mortalità è molto elevata (70%) a causa della scarsa ed aspecifica sintomatologia delle fasi iniziali che rende difficile la diagnosi precoce della malattia.
La terapia è chirurgica per gli stadi I-II-III supplementata dalla chemioterapia con i sali di platino da solo o in associazione. La chemio è la terapia di prima scelta per ca. ovarico al IV° stadio. Nonostante i progressi raggiunti dalla terapia, esiste ancora un’alta percentuale di recidive a causa di una sempre maggiore chemioresistenza (6-10).
Incidenza: Il cancro dell’ovaio colpisce la popolazione femminile in una percentuale di 1.7%. L’incidenza varia a livello internazionale, con i tassi più alti in Nord America e Nord Europa e i più bassi in Giappone, Africa e nei paesi in via di sviluppo. I tassi di incidenza età-specifici mostrano che è piuttosto raro prima dei 40 anni, aumenta dopo i 40 e raggiunge i valori massimi tra i 50 e 69 anni (1-6). In Italia il tasso d’incidenza standardizzato relativo ai dati dei Registri Italiani Tumori negli anni 1993-1998 è pari a 1,3% (1-3). E’ la causa principale (5%) di morte per neoplasie ginecologiche negli Stati Uniti e nei Paesi Occidentali. Muoiono più donne per questo tipo di neoplasia che per il carcinoma della cervice e dell’endometrio considerati insieme. Risulta al 5° posto per frequenza fra tutte le neoplasie maligne dopo il cancro al pancreas, colon, stomaco e retto ed è in modesta regressione negli ultimi 10 anni. Negli ultimi anni si assiste ad un lieve decremento dell’incidenza e della mortalità per cancro ovaio specialmente nell’Italia meridionale (48,49) da attribuire probabilmente all’aumentato utilizzo dei contraccettivi orali e alla migliorata organizzazione ospedaliera.
EZIOLOGIA: l’eziologia e la patogenesi di questa neoplasia non sono ancora ben definiti. Studi epidemiologici indicano incidenze maggiori nelle pazienti con disfunzione ovarica (PCOS), nelle nullipare, nelle pazienti con anamnesi di poliabortività, e nelle pz. che utilizzano farmaci induttori dell’ovulazione come il clomifene e le gonadotropine. Anche l’obesità ed il fumo sembrano avere un ruolo nell’etiopatogenesi del cancro ovarico. La teoria dell“ovulazione incessante” di Fathalla sembra essere l’elemento fondamentale del processo etiologico del cancro ovarico che prevede un’anomalia nel processo di riparazione dello stigma ovulatorio sulla superficie ovarica (50). Se durante la vita una donna ha molte ovulazioni, poche gravidanza e periodi di allattamento molto brevi, ha un rischio maggiore di cancro ovarico. La terapia estrogenica sostitutiva (HRT) in menopausa sembra incrementare il rischio di cancro ovarico in caso di somministrazione HRT >11 anni (20-22).
Ogni gravidanza riduce il rischio di cancro ovarico di circa il 10% (209) e anche l’allattamento e la sezione e legatura delle tube sembrano ridurre il rischio di ca. ovarico. I contraccettivi orali, tramite il loro effetto ovariostatico, riducono il rischio di cancro ovarico dell’11% per ogni anno di terapia sia nelle pazienti con anamnesi familiare oncologica positiva sia nella popolazione generale (210).
Eziologia genetica: Le principali sindromi genetiche correlate al carcinoma ovarico attualmente conosciute sono la sindrome del carcinoma mammario-ovarico (HBOC), la sindrome specifica del carcinoma ovarico e la sindrome di Lynch tipo II. Accenneremo inoltre ad altre sindromi meno importanti correlate al cancro ovarico.
- la sindrome del carcinoma mammario-ovarico (HBOC), legata a una mutazione ereditaria dei geni BRCA-2 e soprattutto BRCA-1, responsabile del 12% delle neoplasie maligne dell’ovaio (95-97). Questa sindrome è caratterizzata dal riscontro in numerosi membri della stessa famiglia affetti di
carcinoma della mammella e/o dell’ovaio (tipicamente 5 o più casi in familiari di primo e secondo grado) o almeno 3 casi di carcinoma mammario od ovarico a insorgenza precoce, cioè prima dei 50 anni di età. nelle donne con forme ereditarie di cancro ovarico-mammario. Il locus di suscettibilità della BRCA-1 è localizzato sul cromosoma 17q12-21 mentre il gene BRCA-2 è localizzato sul cromosoma 13q12-13 (98). E’ probabile che BRCA-1 sia un gene oncosoppressore e la proteina sintetizzata agisca come regolatore negativo della crescita tumorale (3). E’ stato descritto un gran numero di mutazioni di BRCA-1. Nella maggioranza dei casi si tratta di mutazioni “frameshift” o senza senso e nell’86% dei casi si ha la produzione di proteine troncate (46,47). Tra le donne ebree Ashkenaziti, discendenti da gruppi originari dell’Europa Centro-Orientale, la stima dei portatori di mutazioni founder a carico dei geni BRCA1 e BRCA2 è circa del 2,5% (99-120). Le mutazioni dei geni BCRA-1 e 2 possono causare inoltre l’insorgenza dell’anemia di Falconi
E’ probabile che BRCA-1 sia un gene oncosoppressore e la proteina sintetizzata agisca come regolatore negativo della crescita tumorale (3). E’ stato descritto un gran numero di mutazioni di BRCA-1. Nella maggioranza dei casi si tratta di mutazioni “frameshift” o senza senso e nell’86% dei casi si ha la produzione di proteine troncate (46,47). Tra le donne ebree Ashkenaziti, discendenti da gruppi originari dell’Europa Centro-Orientale, la stima dei portatori di mutazioni founder a carico dei geni BRCA1 e BRCA2 è circa del 2,5% (99-120). Le mutazioni dei geni BCRA-1 e 2 possono causare inoltre l’insorgenza dell’anemia di Falconi 
- la sindrome specifica del carcinoma ovarico: 10-15% dei casi
- la sindrome di Lynch tipo II, che può includere i carcinomi del colon, della mammella, dell’endometrio, del primo tratto gastrointestinale e dell’ovaio ed è generalmente associata a una storia familiare di neoplasie a comparsa precoce.
- Sindrome di Cowden: rara malattia ereditaria, autosomica dominante. Fu descritta per la prima volta nel 1963 e prese il nome dalla prima famiglia nella quale la sindrome fu studiata. E’ dovuta ad una mutazione del gene oncosoppressore PTEN (Phosphatase and Tensin homolog) situato nel cromosoma 10 nel locus 10q23.2. La funzione di PTEN è quella di modulare correttamente i processi della via PI3K/Akt/mTOR che governa la proliferazione cellulare e l’apoptosi (139-169). Quasi sempre si ritrova un’associazione tra tumori ovarici e tumori cerebrali;
- Sindrome di Gorlin anche nota come sindrome del carcinoma nevico delle cellule basali (Nevoid Basal Cell Carcinoma Syndrome – NBCCS), è una malattia ereditaria caratterizzata da un’ampia
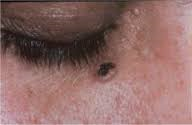
gamma di anomalie dello sviluppo e dalla predisposizione alle neoplasie. L’NBCCS è causata dalle mutazioni del gene PTCH1 e si trasmette con modalità autosomica dominante a penetranza completa e espressività variabile. I raggi solari ultravioletti (UV) sono i principali agenti responsabili di questi tumori, e le persone con pelle chiara sono particolarmente a rischio. I NBCC sorgono in piccoli numeri sulla pelle esposta al sole specialmente nelle persone >50 anni, anche se i più giovani possono anche essere colpiti. NBCCS ha un’incidenza di 1 su 50.000-150.000 con una maggiore incidenza in Australia. NBCC possono comparire anche in aree del corpo che generalmente non sono esposti alla luce solare, come le palme delle mani e le piante dei piedi (176-178).

- La sindrome di Peutz Jeghers (poliposi intestinale amartosica e pigmentazione muco-cutanea), sindrome autosomica dominante caratterizzata geneticamente da una mutazione del gene oncosoppressore STK11 situato sul cromosoma 19, si associa ai tumori dei cordoni stromali sessuali dell’ovaio, piccoli, multifocali e bilaterali: S.C.T.A.T. (Tumore dei Cordoni Sessuali con Tubuli Anulari).
- Alterazioni del gene CDK12 (Cyclin-Dependent Kinase 12): Il complesso Cdk12 / CycK è un oncosoppressore in quanto promuove l’espressione di un sottoinsieme di geni RNA polimerasi II, compresi quelli che dovrebbero provvedere al mantenimento della stabilità genomica e alla riparazione del DNA danneggiato. Ovviamente tale funzione oncosoppressiva non viene esercitata in caso di alterazioni del gene CDK12 con conseguente incremento del rischio oncologico soprattutto a carico dell’ovaio (186-193).
- Disgenesia gonadica (genotipo 46XY puro o mosaiforme): sviluppa gonadoblastomi
Familiarità: l’anamnesi familiare positiva è il fattore di maggior rischio (5%) di tutti i tumori maligni ovarici (170,171). Le donne con un familiare di primo grado affetto da ca. ovarico o mammario o del colon hanno un rischio pari al 5% rispetto all’1,7% della popolazione generale di sviluppare un ca. ovarico. Nelle famiglie con due o più familiari di primo grado affetti il rischio può superare il 50% (90-94). Si riconoscono tre tipi di tumori maligni con ricorrenza familiare ed eredità autosomica dominante:
1) Site-specific ovarian cancer syndrome: si evidenzia solo cancro ovarico.
2) Breast and ovarian cancer syndrome: familiare associato al carcinoma della mammella;
3) Hereditary non polyposis colorectal cancer syndrome (HNPPC): sindrome familiare neoplastica che include carcinoma colon-rettale non polipomatoso e cancro ovarico. Questa sindrome costituisce il 10-15% dei casi di tumore ovarico ereditario ed è correlata a mutazioni dei geni MLH1 e MSH2.
Fattori di rischio:
-
- Età: L’incidenza di carcinoma ovarico aumenta con l’età. E’ virtualmente nulla al di sotto dei 20 anni, anche tra le portatrici di mutazioni dei geni BRCA1/2, rimane molto bassa fino ai 30 anni, mentre cresce linearmente nel periodo 40-50 anni e poi con una velocità minore fino all’ottava decade di vita, quando si registra il tasso più alto. Il tasso di incidenza passa da 1,7% 40 anni a 5,4% a 75 anni, e più di un terzo dei carcinomi ovarici vengono diagnosticati in donne di età >65 anni (15).
- fumo: ritenuto un importante fattore di rischio dalla maggior parte degli studiosi che hnno riscontrato un forte rapporto fra il fumo ed un particolare tipo di cancro ovarico. il tumore mucinoide che rappresenta il 15% di tutti i tumori ovarici. Ma alcuni AA. al contrario ritengono il fumo un fattore protettivo dal cancro dell’ovaio (10) come i contraccettivi (11) e la legatura delle tube (10,56).

- Obesità: le cellule adipose producono adipo-chinine che possono promuovere la proliferazione cellulare (leptine) oppure avere effetto inibitorio sulla proliferazione cellulare (adiponectina). Le donne obese si trovano come in uno stato infiammatorio cronico anche se di basso livello. Lo stato flogistico è di per se stesso un fattore favorente la proliferazione cellulare e l’insorgenza di tumori. L’’aumento del peso, specialmente in pre-menopausa, provoca una iperproduzione di FSH, LH ed estrogeni che stimola la proliferazione delle cellule ovariche, un fattore di rischio lieve/moderato per lo sviluppo di cancro alle ovaie (43,44). In menopausa, invece, elevati livelli di androgeni come Androstenedione e DHEA-s e bassi livelli di estrogeni sono fattori di rischio per cancro ovarico in done obese (179-185). Le donne obese, inoltre, tendono ad avere iperinsulinemia, insulino-resistenza e livelli superiori alla media di IGF-1 (Insulin-like Growth Factor 1), ormoni fortemente sospettati di avere legami con alcuni tipi di tumore (50). L’aumento di rischio oncologico è direttamente correlato all’aumento del peso come dimostrato in studi che hanno espresso effetto di un odds ratio (OR), rapporto di rischio, o tasso di incidenza standardizzato e intervallo di confidenza 95% (CI). Non vi era alcuna prova che l’associazione varia per i diversi sottotipi istologici di tumore di ovarico (179-185).
- Nulliparità: in base alla teoria dell’ovulazione incessante di Fathalla, se durante la vita una donna ha tante ovulazioni (quindi poche gravidanza e brevi periodi di allattamento) ha un rischio maggiore di tumore alle ovaie.
- Induttori dell’ovulazione: l’impiego di terapie ormonali per indurre l’ovulazione sembra associato a un aumento del rischio di carcinoma ovarico tra la popolazione generale e tra le pazienti con una predisposizione genetica al carcinoma ovarico. In studi collaborativi caso-controllo si è riscontrato un aumento del rischio di 3 volte (OR 2,8 IC 95% 1,3-6,1) ed è sostanzialmente maggiore fra le nulligravide (OR 27,0) (24). Data la scarsità di studi prospettici, la non concordanza fra i diversi studi caso-controllo e la presenza di alte percentuali di pazienti PCOS fra le donne esaminate, questi risultati devono essere ulteriormente confermati (25).
- Terapia ormonale sostitutiva (HRT) prolungata: l’utilizzo di terapie estrogeniche in menopausa appare associato a un lieve aumento del rischio di carcinoma ovarico. Una metanalisi di 9 studi ha evidenziato un aumento di incidenza pari al 15% per le donne che avevano utilizzato terapie estrogeniche e pari al 25% per quelle che lo avevano fatto per più di 10 anni (20). In questi due gruppi di pazienti anche il rischio di morte da carcinoma ovarico era superiore rispetto a quello di donne che non avevano mai utilizzato HRT (RR 1,51 e RR 2,20 rispettivamente) (livello III). Tale aumento di rischio non sembra esistere tra le pazienti che assumono terapie combinate estroprogestiniche, sebbene i dati siano insufficienti a trarre conclusioni su questo punto (21). Non esistono dati sull’effetto della HRT nelle pazienti con una predisposizione genetica al carcinoma ovarico (22,51).
- Endometriosi (53,54)
- Familiarità o precedente di cancro alle ovaie, al seno o del colon-retto. Le pz. affette da ca. ovarico con anamnesi familiare positiva rappresentano il 5% di tutte le pz. affette da ca. ovarico. Nelle famiglie con due o più familiari di primo grado affetti da ca. ovarico, il rischio di sviluppare il ca. ovarico è del 20-50% rispetto all’1.7% della popolazione generale (16,17).
- Esposizione perineale al talco in polvere o diaframma vaginale contenente talco
- HNPCC (Hereditary Non-Polyposis Colon Cancer), conosciuta anche come Sindrome di Lynch, è una malattia tumorale autosomica dominante caratterizzata da due manifestazioni fenotipiche: La sindrome di Lynch I, che è caratterizzata dall’insorgenza di una neoplasia al colon ad un’età media di circa 45 anni. La sindrome di Lynch II, che oltre al tumore al colon comprende lo sviluppo di neoplasie extracoloniche, a livello dell’endometrio, dell’ovaio, dello stomaco, del tratto urinario, dei dotti biliari.
Modificazione dei fattori di rischio:
Classificazione istologica (OMS 1999):
- Tumori delle cellule epiteliali, originano da cellule sulla superficie delle ovaie; rappresentano il (90%) dei tumori ovarici.
- Tumori delle cellule germinali, iniziano nelle cellule che producono gli ovociti. Essi possono essere benigni o maligni. La maggior parte sono benigni.
- Tumori stromali o dei cordoni sessuali, nascono nelle cellule dello stroma ovarico.
1) TUMORI OVARICI EPITELIALI (85%): si formano nelle donne con un età compresa tra 20 e 60 anni circa con una frequenza del 60-70%. Si distinguono cinque sottotipi principali: sierosi (50%), mucinosi (25%), endometriodi (15%), a cellule chiare (5%), tumore di Brenner (1%) e indifferenziati (4).
A) TUMORI OVARICI SIEROSI:
- cistoadenoma sieroso ovarico: 60% dei tumori sierosi,
- cistoadenoma sieroso ovarico borderline: 15% dei tumori sierosi,
- cistoadenocarcinoma sieroso ovarico: 25% dei tumori sierosi, è il tipo più frequente di cancro dell’ovaio.
B) TUMORI OVARICI MUCINOSI: 20% di tutti i tumori ovarici
- cistadenoma mucinoso ovarico: 80% dei tumori mucinosi,
- cistadenoma mucinoso ovarico borderline: 10-15% dei tumori mucinosi. Tumore borderline vuol dire intermedio tra benigno e maligno,
- cistadenocarcinoma mucinoso ovarico: 5-10% dei tumori mucinosi.
C) TUMORI OVARICI ENDOMETRIOIDI: 8-15% di tutti i tumori ovarici
- cistoadenofibroma ovarico, a volte classificato come una categoria separata, piuttosto che sotto gli epiteliali,
- adenofibroma ovarico: può essere sieroso, mucinoso, endometrioide, a cellule chiare o misto,
- cistoadenocarcinofibroma ovarico: estremamente raro.
D) CARCINOMA OVARICO A CELLULE CHIARE: 5% di tutti i cancri ovarici.
E) TUMORI DI BRENNER: 2-3% delle neoplasie epiteliali dell’ovaio, sono generalmente benigni e monolaterali.; interessano le donne fra i 30-70 anni con un picco fra i 40-60 anni.
I tumori di Brenner sono di tipo misto stroma-epiteliale; l’epitelio è di tipo transizionale, simile all’urotelio, epitelio della mucosa vescicale, e sono suddivisi in:
- Tumore benigno di Brenner (fibroma)
- Tumore di Brenner borderline (a basso potenziale maligno)
- Tumore maligno di Brenner
- Carcinoma a cellule transizionali.
I tumori benigni e borderline sono asintomatici e la diagnosi è spesso occasionale, emerge infatti durante indagini diagnostiche per altre patologie; la prognosi è eccellente. I tumori di Brenner maligni invece , si manifestano in modo analogo agli altri tipi di cancro ovarico come massa addominale e pelvica con sintomatologia correlata a disturbi di tipo gastrointestinale e urologico.
Macroscopicamente i tumori a cellule di Brenner si presentano come neoplasie con diametro >20 cm. Le forme benigne e bordeline sono costituite da lesioni con superficie esterna liscia e al taglio sono generalmente uniloculate; le forme maligne hanno superficie esterna irregolare e al taglio sono presenti aree solide, necrotiche ed emorragiche. Istologicamente la neoplasia è composta da epitelio uroteliale. Le forme benigne e borderline mostrano lievi e rare atipie delle cellule; le forme maligne sono caratterizzate da papille rivestite da epitelio atipico, con aree di necrosi ed emorragia. Vi possono essere inoltre aree composte da cellule che producono muco.
2) TUMORI DELLE CELLULE GERMINALI DELL’OVAIO (5% delle neoplasie ovariche): derivano dalle cellule germinali che danno origine agli ovociti. Interessano quasi esclusivamente le donne <30 anni e rappresentano il 5% dei cancri ovarici. Se il tumore ha le caratteristiche della cellula germinale che ancora non è stata fecondata, abbiamo il disgerminoma. Se il tumore ha le caratteristiche della cellula germinale fecondata che si differenzia in senso embrionale, abbiamo il carcinoma embrionale e i teratomi. Se il tumore ha le caratteristiche delle cellule germinali progenitrici della placenta e delle membrane amnio-coriali, si ha il chorioncarcinoma e il tumore del sacco vitellino (o tumore del seno endodermico).
Disgerminoma: il 10-20% di questi tumori insorge durante la gravidanza. Clinicamente si manifesta con una massa addominale e/o pelvica a rapida crescita e con aumento dei livelli sierici di LDH (lattico deidrogenasi) e PLAP (fosfatasi alcalina placentare). La prognosi dipende dallo stadio della malattia.
Macroscopicamente il tumore è una massa solida che può raggiungere i 15 cm di diametro e ha margini lobulati; al taglio è di colore rosso-giallastro con calcificazioni. Nel 10-15% dei casi vi è coinvolgimento bilaterale delle ovaie. Istologicamente il tumore è formato da cellule grandi che si dispongono in nidi e cordoni; commiste vi sono cellule infiammatorie e calcificazioni. All’analisi citogenetica sono presenti anomalie del cromosoma 12.
Carcinoma embrionario: tumore raro dell’ovaio (3% dei tumori germinali dell’ovaio). Le cellule tumorali somigliano alla cellula uovo che è stata fecondata e dunque il tumore è composto da tessuti che somigliano all’embrione. Insorge nelle giovani donne in età prepuberale e si presenta come una massa pelvica e/o addominale; la sintomatologia è legata alla produzione da parte delle cellule tumorali di un ormone, la β-hCG, che causa pubertà precoce, alterazioni del ciclo mestruale e talora sintomatologia gravidica (nausea, vomito). La prognosi è legata allo stadio della malattia ma nel 50% dei casi la diagnosi è effettuata in fase tardiva con presenza di metastasi. Macroscopicamente il tumore è una neoplasia solida monolaterale, con diametro >15 cm; al taglio si presenta di colorito grigiastro con aree di necrosi ed emorragia. Istologicamente il tumore è composto da cellule di grandezza variabile che si dispongono a formare ghiandole o papille frammiste ad aree di necrosi. Possono essere presenti nidi di cellule che somigliano al sacco vitellino o al disco placentare. La terapia è chirurgica e chemioterapica.
Teratomi: I teratomi sono tumori composti da tessuti che somigliano a quelli di un piccolo feto e si suddividono in maturi, immaturi e “specializzati”.
Teratoma maturo (o cisti dermoide dell’ovaio): sono tumori benigni che si generano dalla fusione anomala di due cellule germinali (partenogenesi) e costituiscono il 20-40% di tutti i tumori ovarici e più del 95% dei tumori a cellule germinali. Clinicamente sono tumori a lenta crescita, asintomatici e insorgono nelle donne di età compresa tra i 20 e i 40 anni. La prognosi è eccellente. Macroscopicamente sono lesioni monolaterali del diametro <10 cm, a superficie polilobulata. Al taglio è presente materiale pilo-sebaceo e sulla superficie interna sono presenti nidi solidi contenenti denti e osso. Istologicamente il tumore è composto da tutti i tessuti che compongono il corpo umano e quindi possiamo vedere, in varia percentuale, tessuto tiroideo, intestinale, polmonare, tessuto nervoso, epatico e altro.
Teratoma specializzato: variante caratterizzata a livello istologico dalla presenza di un solo tessuto. Nella maggior parte dei casi il tessuto rappresentato è quello tiroideo e si parla infatti di “struma ovarii”. Altri teratomi specializzati sono i carcinoidi (che insorgono in età adulta e sono associati a produzione di ormoni e causano dunque sindromi endocrine) e i tumori neuro-ectodermici (formati esclusivamente da tessuto nervoso).
Teratoma immaturo: variante caratterizzata dalla presenza di tessuti che sono “immaturi”, cioè non ben sviluppati. Si presentano come masse addominali e clinicamente con elevati livelli sierici di AFP. La prognosi è eccellente nei primi stadi di malattia. Macroscopicamente sono tumori di alcuni centimetri a consistenza solida e al taglio si presentano cistici. Istologicamente, a seconda della percentuale di tessuto immaturo che viene identificato, i tumori si classificano in Grado 1 (poco tessuto immaturo), grado 2 (tessuto immaturo in quantità moderata) e grado 3 (tessuto immaturo in quantità elevata).
- teratoma ovarico: il più comune tumore benigno primario dell’ovaia,
- teratoma ovarico maturo,
- teratoma ovarico immaturo,
- teratomi specializzati dell’ovaio,
- Chorion-carcinoma: Il choriocarcinoma è un tumore che si può sviluppare in assenza o in presenza di gravidanza. Il chorion-carcinoma non gravidico è un tumore ad alto grado di malignità che rappresenta meno dell’1% di tutti i tumori a cellule germinali. Nella maggior parte dei casi si presenta come parte di un tumore misto. Il chorion-carcinoma ovarico primario puro è estremamente raro, insorge dalla placenta e metastatizza all’ovaio. Il choriocarcinoma colpisce le donne giovani, <20 anni, e si presenta come una massa addominale e /o pelvica a rapida crescita che può provocare emoperitoneo e manifestazioni endocrine dovute alla produzione di β-hCG. Spesso al momento della diagnosi sono presenti metastasi polmonari. La prognosi non è buona e comunque dipende dallo stadio della malattia. Macroscopicamente il tumore è una lesione del diametro di alcuni centimetri, emorragica al taglio. Istologicamente è formata da cellule sinciziali placentari, commiste a vaste aree di necrosi e di emorragia.
Carcinoma del sacco vitellino: per frequenza è il secondo tumore a cellule germinali maligno più frequente (20% di tutte le neoplasie a cellule germinali). Interessa specialmente le bambine e le giovani donne. Si presenta come un tumore a rapida crescita che determina dolore addominale; clinicamente. E’ presente aumento dell’AFP (alfa-feto-proteina) e del CA-125. La prognosi è eccellente se il tumore viene diagnosticato agli stadi iniziali. Macroscopicamente il tumore è una lesione monolaterale, del diametro di alcuni centimetri di colorito grigiastro. Istologicamente il tumore è formato da cellule disposte in microcisti e piccole papille e che talora formano ghiandole che somigliano a quelle presenti nell’intestino o nell’endometrio e, caratteristici, sono i corpi di Schiller-Duval, formazioni simili ai glomeruli. All’esame citogenetico sono presenti alterazioni del cromosoma 12.
- Tumore maligno misto a cellule germinali dell’ovaio
3. TUMORI STROMALI OVARICI: 8-10% DELLE NEOPLASIE OVARICHE
I tumori stromali ovarici derivano dalle cellule stromali che circondano l’ovocita. Ci sono poi altre cellule stromali poco sviluppate nelle donne, le cellule di Sertoli-Leydig, che sono predominanti invece nella gonade maschile e che hanno anche attività endocrina.
- fibroma ovarico: 4% dei tumori ovarici
- tecoma ovarico: 5% dei tumori ovarici,
- Tumore stromale sclerosante dell’ovaio
- Tumore a cellula della granulosa
- Tumore a cellule di Sertoli-Laydig
- Carcinosarcoma ovarico: <1%
Fibroma ovarico
Il fibroma è una neoplasia benigna, tipicamente monolaterale, e costituisce meno del 10% delle neoplasie primitive dell’ovaio. Insorge dalla quarta decade di vita e può essere asintomatico e diagnosticato accidentalmente durante esami condotti per altre patologie, o manifestarsi con dolore addominale-pelvico, causato dalla torsione sul suo asse. Macroscopicamente è una lesione nodulare del diametro variabile da pochi millimetri a qualche centimetro, a margini netti e di consistenza solida. Al taglio è bianco ed omogeneo, talora con aree cistiche. Istologicamente è formato da cellule fusate, tutte uguali tra loro. La prognosi è eccellente e la chirurgia è risolutiva.
I fibromi ovarici possono insorgere nelle pazienti con sindrome di Gorlin; in questo caso sono bilaterali e si manifestano prima dei 30 anni. Questa sindrome è caratterizzata da tumori della pelle, anomalie scheletriche e altri tumori che si manifestano con l’aumentare dell’età.
Tecoma ovarico
Il tecoma è composto da cellule adipose che somigliano alle cellule della teca ovarica e da altre cellule stromali. Insorge in post-menopausa e può essere associato a neoplasie dell’endometrio. Si presenta come massa addominale o viene diagnosticato dopo indagini per sanguinamento vaginale causato dal tumore dell’endometrio. Nel 10% dei casi alcune cellule del tumore producono androgeni e dunque il tumore provoca una “sindrome virilizzante”, caratterizzata irsutismo, amenorrea, abbassamento del timbro della voce. Questi tumori sono di per sé benigni e la chirurgia è risolutiva. Macroscopicamente il tecoma si presenta come massa tondeggiante di colorito giallo (per l’abbondante presenza di lipidi) e al taglio è solido con focali aree di emorragia. Nelle donne giovani sono frequenti ed estese le aree calcifiche. Istologicamente il tumore è formato da cellule tonde (simili a quelle della teca) e da cellule fusate, in varia proporzione.
Una rara variante è il tecoma luteinizzato con peritonite sclerosante, che colpisce le donne nella terza e nella quarta decade di vita. L’eziologia è ancora sconosciuta; in questa entità il tecoma si associa ad alterazioni del peritoneo (peritonite sclerosante). Il peritoneo si ispessisce e determina alterazione della funzionalità dell’intestino, causando pseudo occlusioni e dolori addominali. A volte è colpita anche la pleura, e si genera versamento pleurico. In questo caso, sono affette da tecoma entrambe le ovaie e la neoplasia può raggiungere anche i 30 cm di diametro.
Tumore stromale sclerosante dell’ovaio
È una rara neoplasia benigna che costituisce circa il 2-6% di tutti i tumori stromali dell’ovaio, insorge in età giovanile e la prognosi è eccellente. Nella maggior parte dei casi è monolaterale. Macroscopicamente è una neoplasia a margini lobulati e a superficie esterna liscia, con diametro che può raggiungere anche i 20 cm. Al taglio il tumore è delimitato da tessuto ovarico residuo mentre la parte centrale può essere cistica. Tra la parte centrale cistica e la parte esterna costituita da ovaio residuo, vi è una parte solida di colorito giallastro che può contenere aree emorragiche. Istologicamente la neoplasia è formata da cellule che si dispongono a nidi e da vasi interposti.
Tumori dei cordoni sessuali: I tumori dello stroma gonadico (cellule di Leydig) e/o dei cordoni sessuali (cellule di Sertoli) rappresentano circa l’8% dei tumori ovarici e si sviluppano nelle cellule del tessuto stromale dell’ovaio. Dato che queste cellule sono ormono-secernenti, si riscontreranno alterazioni ormonali da iperproduzione steroidea. All’interno di questo gruppo, i tumori considerati maligni sono il tumore a cellule della granulosa che rappresenta il più comune tumore maligno dei cordoni sessuali e l’androblastoma o tumore delle cellule di Sertoli-Leydig che si presenta con una frequenza dello 0.5% fra tutti i tumori ovarici. Altri tumori maligni dei cordoni sessuali sono quelli con tubuli anulari (S.C.T.A.T.) che si riscontrano frequentemente nella sindrome di Peutz-Jeghers, i tumori stromali indifferenziati e i fibrosarcomi. I tumori ovarici S.C.T.A.T. (Tumore dei Cordoni Sessuali con Tubuli Anulari) sono quasi sempre piccoli, multifocali, bilaterali e benigni ma possono ritrovarsi forme maligne.
Tumore a cellule della granulosa dell’età adulta (>18 anni): può manifestarsi in ogni età >18 anni ma raggiunge il picco nelle donne in peri- e post-menopausa. Le cellule che lo compongono producono estrogeni e dunque causano iperestrogenismo e conseguente menometrorragia, lesioni endometriali e carcinoma dell’endometrio. La sintomatologia è collegata alla massa addominale o pelvica o ad emoperitoneo provocato da rottura della neoplasia. Nel 95% dei casi il tumore è monolaterale e a bassa potenzialità maligna, ma facile alle recidive. La prognosi dipende dallo stadio al momento della diagnosi. Macroscopicamente il tumore si presenta come una lesione nodulare del diametro di circa 10 cm, ma che può raggiungere i 30 cm, con superficie irregolare. Al taglio si osserva alternanza di aree cistiche e solide ed emorragiche. Istologicamente le cellule tumorali della granulosa si dispongono in nidi, in cordoni o in piccole papille e possono essere commiste a cellule fusate.
Tumore a cellule della granulosa giovanile: rappresenta il 10% dei tumori dell’ovaio insorti nelle donne di età <20 anni. La crescita è aggressiva. La prognosi è legata allo stadio della malattia al momento della diagnosi. Questo tumore, come la controparte a insorgenza in età adulta, produce estrogeni e dunque spesso si manifesta con pubertà precoce. Altri sintomi sono masse addomino-pelviche, dolore addominale, emoperitoneo. L’aspetto macroscopico e istologico è sovrapponibile al tumore a cellule della granulosa dell’adulto. La terapia, in caso di lesione monolaterale, prevede ovariectomia, controllo controlaterale e prelievo bioptico endometriale. Invece, in caso di localizzazione bilaterale o monolaterale con rottura della capsula, occorre procedere con annessiectomia bilaterale + isterectomia totale intrafasciale + polichemioterapia.
Tumori a cellule di Sertoli-Leydig
Questi tumori sono formati dalle cellule del Sertoli-Leydig, che producono ormoni maschili. Le cellule di Leydig secernono testosterone e diidrotestosterone. Le cellule di Sertoli secernono inibina e convertono il testosterone prodotto dalle cellule di Leydig in DHT ed estradiolo. Questi tumori hanno il picco di incidenza intorno ai 25 anni; le forme meno differenziate e quindi più aggressive colpiscono un’età inferiore, le forme più differenziate e quindi meno aggressive colpiscono le donne in età avanzata. Pur derivando da cellule che secernono ormoni, non sempre questi tumori li producono in abbondanza e spesso i primi stadi della malattia sono asintomatici. Solo un terzo delle pazienti si presenta con sindromi virilizzanti.
Esistono 5 categorie istologiche di tumori a cellule di Sertoli-Leydig:
-Tumore a cellule di Sertoli-Leydig ben differenziato,: colpisce le donne di 20-40 anni; è raro e presenta un’architettura tubulare, con tubuli spesso aperti, pervi e più raramente solidi. composti da cellule Sertoli-like alternate a bande di stroma contenenti cellule di Leydig.
–Tumore a cellule di Sertoli-Leydig moderatamente differenziato;
-Tumore a cellule di Sertoli-Leydig scarsamente differenziato, che colpisce le bambine e le adolescenti;
–Variante retiforme, caratterizzata da una crescita con aree cistiche alternate ad aree solide, che fanno somigliare il tumore alla rete testis del testicolo
–Variante con elementi eterologhi, caratterizzati dalla presenza di tessuti di altri organi (esempio, con componente di tipo gastro-intestinale).
Macroscopicamente il tumore appare come una neoplasia ovoidale con superficie irregolare, con diametro maggiore che può raggiungere i 15 cm. Al taglio vi è alternanza di aree solide e cistiche. Queste caratteristiche sono comuni a tutte le varianti istologiche. La prognosi del tumore è strettamente correlata allo stadio della malattia. Due ulteriori varianti sono:
–Tumore a cellule del Sertoli puro, composto cioè solo da cellule del Sertoli. Questo tumore, che ha una buona prognosi, insorge nelle donne in età riproduttiva e si può manifestare con sindromi virilizzanti. Può insorgere inoltre nell’ambito della sindrome di Peutz-Jeghers (pigmentazione delle mucose, polipi amartomatosi -composto da elementi di varia origine embrionale, i quali sono comunque, da un punto di vista istologico, dei tessuti normali- del tratto gastro-intestinale, rari carcinomi del tratto gastrointestinale e adeno-carcinoma della cervice uterina).
Carcino-sarcoma ovarico: Il carcino-sarcoma dell’ovaio è una neoplasia dell’età avanzata (>60  anni) che costituisce l’1% di tutte le neoplasie maligne di quest’organo.
anni) che costituisce l’1% di tutte le neoplasie maligne di quest’organo.
Trattasi di neoplasia mista o bifasica composta da elementi epiteliali e mesenchimali maligni ad alto grado. nella componente carcinomatosa è rappresentata da tessuto sieroso o endometrioide o misto. La componente sarcomatosa si distinguono elementi mulleriani (fibrosarcomi, leiomiosarcomi e sarcoma endometriale stromale) ed elementi non mulleriani (rabdomiosarcomi, condrosarcomi, osteosarcomi e liposarcomi). Macroscopicamente si presenta come una massa solido-cistica con emorragia e necrosi.
La diagnosi purtroppo avviene quasi esclusivamente in fase avanzata, per cui risulta essere a prognosi infausta (194). L’iperespressione p53 è associata a stadio avanzato e ridotta sopravvivenza (195).
La terapia del carcino-sarcoma ovarico prevede l’exeresi chirurgica citoriduttiva per gli stadi I-II-III FIGO e la chemioterapia con Cisplatino 75 + Taxolo 175 mg/mq,mg/mq o Ifosfamide 5 gr/mq per 6-7 cicli e la sola chemioterapia per lo stadio IV (196).
4) ALTRI:
- Carcinoma indifferenziato dell’ovaio: ~4% di tutti i tumori ovarici
- carcinoma a cellule squamose dell’ovaio
- Linfoma ovarico
- Linfoma primario dell’ovaio
- Coinvolgimento secondario dell’ovaio con linfoma
- Metastasi all’ovaio
- Tumore di Krukenburg: masse ovariche bilaterali derivanti da carcinomi gastrointestinali
- Altre lesioni metastastiche all’ovaio
DIAGNOSTICA
Il cancro ovarico localizzato è generalmente asintomatico. Nella maggioranza delle pazienti con cancro ovarico la diagnosi viene posta casualmente nel corso di visite ginecologiche di routine oppure quando la malattia si è già diffusa oltre la pelvi. La comparsa di dolore addominale, distensione dell’addome e sintomi urinari di solito indicano una fase avanzata della malattia. Tuttavia, l’aumento progressivo di un tumore ovarico localizzato può determinare la comparsa di poliuria e stipsi. Diversamente dal carcinoma della cervice o dal carcinoma endometriale, gli episodi metrorragici sono raramente presenti nelle fasi iniziali del cancro ovarico. La diagnosi precoce della malattia di solito avviene in seguito alla palpazione di una massa annessiale asintomatica nel corso di un’esplorazione pelvica di routine. Tuttavia, la maggioranza delle masse ovariche identificate all’esame obiettivo è rappresentata di cisti benigne funzionali che caratteristicamente scompaiono nell’arco di 2-3 cicli mestruali. Masse annessiali che si presentano in donne prima del menarca o dopo la menopausa sono più frequentemente patologiche. Altre cause di masse annessiali includono fibromi uterini peduncolati, endometriosi, neoplasie ovariche benigne e lesioni infiammatorie dell’intestino.
Sintomatologia: Non ci sono sintomi iniziali e la sintomatologia, che appare solo in fase avanzata, è vaga ed aspecifica. I principali sintomi sono:
- disuria e poliuria
- stipsi
- distensione addominale
- Dolore addominale e pelvico
- stanchezza
- dispnea: è conseguenza di ascite e/o versamento pleurico

- Tosse secca
- lombalgia
- disturbi del ciclo mestruale e metrorragia: da tumori ovarici ormono-secernenti
- Nausea e pirosi gastrica da metastasi addominale e compressione gastrica
- Metrorragie e alterazioni del ciclo sono rari e quasi sempe da attribuire a lesioni ormonosecernenti.
Esame ginecologico: Con l’esplorazione pelvica bimanuale si palpa in sede annessiale una massa ovarica di diametro >3 cm. Prima della menopausa l’ovaio normale è circa di 2,5 cm. Dopo la menopausa si atrofizza e presenta un diametro <2 cm. In una donna fertile è normale riscontrare un ovaio palpabile, ma dopo la menopausa generalmente ciò indica un tumore ovarico, benigno o maligno. Nonostante non esistano studi che abbiano stimato la sensibilità e la specificità della sola esplorazione bimanuale, si ritiene che il potere predittivo di questa metodica sia basso e gravato da un alto rischio di falsi positivi, specie in epoca premenopausale per la maggiore prevalenza di masse annessiali di natura funzionale (13).
CA-125: I livelli di questa proteina tendono ad essere più elevati in alcune donne con cancro ovarico. I determinanti del CA-125 sono glicoproteine con dimensioni molecolari comprese tra 220 e 1000 kDa. Circa l’80-85% delle pazienti con cancro ovarico presenta livelli di CA- 125 ≥35 U/ml. Purtroppo il 50% circa delle pz. con ca. ovarico allo stadio I-II presenta livelli di CA-125 <65 U/ml. Anche altri tumori maligni possono dar luogo a livelli elevati di CA-125, come i carcinomi dell’endometrio, della cervice, della tuba, del pancreas, della mammella, del polmone e del colon (1). Condizioni non maligne talvolta caratterizzate da valori elevati di CA125 comprendono la gravidanza, l’endometriosi, la malattia infiammatoria pelvica (PID) e i fibromi uterini. Circa 1% delle donne normali ha livelli sierici di CA-125 >35 U/ml. Comunque, in donne in età postmenopausale con una massa pelvica asintomatica e livelli di CA125 ≥65 U/ml la sensibilità del test è del 97% e la sua specificità del 78%. Il dosaggio sierico di CA 125 non è sufficiente per effettuare uno screening di massa per evidenziare il cancro dell’ovaio in fase precoce; nenche associato con l’ETV.
Altri markers: CA 19.9; CA 15.3; CEA; alfa-FP; beta-HCG; LDH
Mutazioni del gene BRCA-1 e BRCA-2: dai leucociti viene estratto il DNA su cui viene eseguita un sequenziamento completo degli esoni codificanti i geni BRCA1 e BRCA2.
Dosaggio sierico di FSH, LH, Androstenedione, DHEA-s: bassi livelli sierici di gonadotropine e elevate concentrazioni di androgeni costituiscono un incrementato rischio di cancro ovarico (45)
Ecografia transvaginale (ETV): ha sostituito l’ecografia addominale, più lenta e meno sensibile, ma presenta  un significativo numero di risultati falsamente positivi, in particolare modo nelle donne in età premenopausale. L’USG in grado di stimare le dimensioni dell’ovaio, di rilevare masse piccole fino ad 1 cm, e distinguere le lesioni solide dalle liquide. Il color Doppler consente di rilevare la presenza di vascolarizzazione interna alla massa in scansione ed associazione con l’ecografia transvaginale può migliorare l’accuratezza e ridurre l’elevato rischio di risultati falsamente positivi: la percentuale di falsi positivi è maggiore (1-2,5%) per l’ecografia che per il Ca125 (0,1-0,6%). Dal punto di vista morfologico il ca. ovarico può presentarsi con aspetto monoloculare, con aspetto monoloculare ed iperecogenicità inteme, sotto forma di cisti biloculari o multiloculari, tumori cistici con aspetti solidi e tumori con aspetti solamente solidi. le cisti monoloculari semplici possono presentare un tumore maligno nello 0,5% dei casi; le cisti multiloculari presentano l’I% dei casi di malignità; le cisti con aspetti solidi
un significativo numero di risultati falsamente positivi, in particolare modo nelle donne in età premenopausale. L’USG in grado di stimare le dimensioni dell’ovaio, di rilevare masse piccole fino ad 1 cm, e distinguere le lesioni solide dalle liquide. Il color Doppler consente di rilevare la presenza di vascolarizzazione interna alla massa in scansione ed associazione con l’ecografia transvaginale può migliorare l’accuratezza e ridurre l’elevato rischio di risultati falsamente positivi: la percentuale di falsi positivi è maggiore (1-2,5%) per l’ecografia che per il Ca125 (0,1-0,6%). Dal punto di vista morfologico il ca. ovarico può presentarsi con aspetto monoloculare, con aspetto monoloculare ed iperecogenicità inteme, sotto forma di cisti biloculari o multiloculari, tumori cistici con aspetti solidi e tumori con aspetti solamente solidi. le cisti monoloculari semplici possono presentare un tumore maligno nello 0,5% dei casi; le cisti multiloculari presentano l’I% dei casi di malignità; le cisti con aspetti solidi  possono presentare il 5-6% dei casi di malignità; le formazioni solide possono presentare il 9% dei casi di malignità. Le componenti solide, le papille, i setti ispessiti sono correlati significativamente con le neoplasie ovariche maligne, incrementando con il loro rilevamento la sensibilità e la specificità dell’indagine ecografica transvaginale.Tra gli aspetti morfologici ecografici di sospetto ricordiamo le formazioni cistiche di dimensioni >5 cm; cisti con pareti leggermente irregolari; presenza di ecogenicità o di setti sottili; presenza di aspetti solidi di volume incrementale; aspetti multinodulari con presenza di setti multipli; presenza di ascite. Segni ecografici di malignità: presenza di multilocularità, setti ispessiti, aree di solidità ed aspetti multicistici, pareti irregolari, proliferazioni aggettanti in cavità. L’altro aspetto valutativo ed indicativo di malignità
possono presentare il 5-6% dei casi di malignità; le formazioni solide possono presentare il 9% dei casi di malignità. Le componenti solide, le papille, i setti ispessiti sono correlati significativamente con le neoplasie ovariche maligne, incrementando con il loro rilevamento la sensibilità e la specificità dell’indagine ecografica transvaginale.Tra gli aspetti morfologici ecografici di sospetto ricordiamo le formazioni cistiche di dimensioni >5 cm; cisti con pareti leggermente irregolari; presenza di ecogenicità o di setti sottili; presenza di aspetti solidi di volume incrementale; aspetti multinodulari con presenza di setti multipli; presenza di ascite. Segni ecografici di malignità: presenza di multilocularità, setti ispessiti, aree di solidità ed aspetti multicistici, pareti irregolari, proliferazioni aggettanti in cavità. L’altro aspetto valutativo ed indicativo di malignità 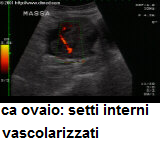 a disposizione dell’indagine ecografica è l’esplorazione color-Doppler. Sappiamo infatti che i fattori di
a disposizione dell’indagine ecografica è l’esplorazione color-Doppler. Sappiamo infatti che i fattori di  crescita tumorali deteminano uno sviluppo rapido e disordinato dei vasi: le pareti vasali appaiono alterate. Tra i fattori neoangiogenetici che appaiono predominare, vi sono quelli della crescita endoteliale: i vasi si manifestano con decorso irregolare e bizzarro, con presenza di shunt artero-venosi; nella parete vasale l’assenza della muscolatura liscia fa sì che le resistenze vascolari siano ridotte. Il decorso si spesso interrompe bruscamente ed, all’esplorazione color, il flusso assume in alcune diramazioni colori molteplici, indicativi della bizzarria della struttura vascolare. La vascolarizzazione tumorale può essere di tipo periferica, centrale, di tipo papillare o settale. Alcuni autori hanno rilevato come la sede settale e papillifera sia significativamente correlabile alla malignità ovarica. Altri studi confermano come la vascolarizzazione tumorale sia più frequentemente distribuita a livello centrale nei tumori maligni rispetto a quelli benigni (65% versus 5%) in cui la distribuzione appare più periferica (aspetti qualitativi). Il notch astolico (segno di alta resistenza) è evidenziabile nell’89% dei tumori benigni ma in nessuno di quelli maligni: il PI è dello 0,56 +/- 0,13 per i tumori maligni e dello 1,06 +/- 0,07 per quelli benigni.
crescita tumorali deteminano uno sviluppo rapido e disordinato dei vasi: le pareti vasali appaiono alterate. Tra i fattori neoangiogenetici che appaiono predominare, vi sono quelli della crescita endoteliale: i vasi si manifestano con decorso irregolare e bizzarro, con presenza di shunt artero-venosi; nella parete vasale l’assenza della muscolatura liscia fa sì che le resistenze vascolari siano ridotte. Il decorso si spesso interrompe bruscamente ed, all’esplorazione color, il flusso assume in alcune diramazioni colori molteplici, indicativi della bizzarria della struttura vascolare. La vascolarizzazione tumorale può essere di tipo periferica, centrale, di tipo papillare o settale. Alcuni autori hanno rilevato come la sede settale e papillifera sia significativamente correlabile alla malignità ovarica. Altri studi confermano come la vascolarizzazione tumorale sia più frequentemente distribuita a livello centrale nei tumori maligni rispetto a quelli benigni (65% versus 5%) in cui la distribuzione appare più periferica (aspetti qualitativi). Il notch astolico (segno di alta resistenza) è evidenziabile nell’89% dei tumori benigni ma in nessuno di quelli maligni: il PI è dello 0,56 +/- 0,13 per i tumori maligni e dello 1,06 +/- 0,07 per quelli benigni.
Metodica Energy: Questa tecnica recente consente una valutazione qualitativa superiore della massa stessa con una maggiore delineazione del percorso vascolare e degli spots (“tumor lakes”). Gli indicatori (aspetti quanfitativi del flusso) utilizzati: l’RI ed il PI calcolati come RI=S-D/S e PI=S-D/Vm. Da notare come il flusso sia a bassissima resistenza. L’indagine ecografica, dunque, consente lo studio degli indici di resistenza vascolari e lo studio della morfologia e dell’estensione vascolare. Nella valutazione degli indicatori i vantaggi sono costituiti dall’avere a disposizione un dato oggettivo riproducibile; tra gli svantaggi, la scelta dell’area di vascolarizzazione da cui trarre l’informazione, quante informazioni si ricavano, a quale informazione dare il valore di cut-off. Nello studio della morfologia e dell’estensione vascolare i vantaggi sono rappresentati dal fatto che questi parametri rappresentano l’espressione biologica del processo neoangiogenetico della massa; tra gli svantaggi i limiti  interpretativi dettati dalla differenza delle apparecchiatura utilizzate e dagli operatori stessi. Tutti gli aspetti interpretativi, sia quantitativi che qualitativi vascolari, si desumono dal significato biologico della neovascolarizzazione come elemento importante del processo neoplastico. Ciò che inficia la validità del color Doppler è, però, l’impossibilità di trovare dei parametri oggettivi e riproducibili nel differenziare il processo neoplastico dagli altri. In altre parole, si è visto che pur utilizzando tutti questi parametri il numero dei falsi positivi è sempre consistente tanto che, operando una donna per sospetta massa ovarica maligna, spesso ci si imbatte in una massa ovarica benigna. Questi vasti overlapping di pattem vascolari fra le forirne benigne e maligne ci ha portato nel tempo a riflettere
interpretativi dettati dalla differenza delle apparecchiatura utilizzate e dagli operatori stessi. Tutti gli aspetti interpretativi, sia quantitativi che qualitativi vascolari, si desumono dal significato biologico della neovascolarizzazione come elemento importante del processo neoplastico. Ciò che inficia la validità del color Doppler è, però, l’impossibilità di trovare dei parametri oggettivi e riproducibili nel differenziare il processo neoplastico dagli altri. In altre parole, si è visto che pur utilizzando tutti questi parametri il numero dei falsi positivi è sempre consistente tanto che, operando una donna per sospetta massa ovarica maligna, spesso ci si imbatte in una massa ovarica benigna. Questi vasti overlapping di pattem vascolari fra le forirne benigne e maligne ci ha portato nel tempo a riflettere sull’identificazione di una variabile che ci conducesse ad un incremento della sensibilità e della specificità della metodica Doppler . Confrontando infatti le donne in età fertile con quelle in epoca postmenopausale si è pensato che si potessero ottenere dei risultati differenti. Le ovaie delle donne in età menopausale appaiono selerotiche, scarsamente vascolarizzate, e quindi qualsiasi aspetto neovascolare può essere suggestivo di malignità. In un nostro studio abbiamo in effetti verificato come il numero dei campionamenti di aree di neovascolarizzazione fosse significativamente superiore nelle donne in epoca postmenopausale rispetto a quelle in età fertile. L’età rappresenta, quindi, un parametro valutativo fondamentale per una più corretta diagnosi nel riscontro istologico. Ricordiamo, infine, che la sola interpretazione delle caratteristiche vascolari è insufficiente e non può prescindere da quella delle caratteristiche morfologiche.
sull’identificazione di una variabile che ci conducesse ad un incremento della sensibilità e della specificità della metodica Doppler . Confrontando infatti le donne in età fertile con quelle in epoca postmenopausale si è pensato che si potessero ottenere dei risultati differenti. Le ovaie delle donne in età menopausale appaiono selerotiche, scarsamente vascolarizzate, e quindi qualsiasi aspetto neovascolare può essere suggestivo di malignità. In un nostro studio abbiamo in effetti verificato come il numero dei campionamenti di aree di neovascolarizzazione fosse significativamente superiore nelle donne in epoca postmenopausale rispetto a quelle in età fertile. L’età rappresenta, quindi, un parametro valutativo fondamentale per una più corretta diagnosi nel riscontro istologico. Ricordiamo, infine, che la sola interpretazione delle caratteristiche vascolari è insufficiente e non può prescindere da quella delle caratteristiche morfologiche.

Tomografia computerizzata (TAC),
RMN con m.d.c.,
Tomografia ad emissione di positroni (PET)
Rx- torace
Laparoscopia: il sospetto di ca. ovarico costituisce una delle principali indicazioni della LPS diagnostica ed operativa per masse ovariche di diametro <8 cm. In caso di dubbio diagnostico si può tranquillamente effettuare una biopsia ovarica con  esame istologico estemporaneo. I
esame istologico estemporaneo. I  più recenti studi oncologici hanno dimostrato che la biopsia ovarica non fa peggiorare la prognosi anche in caso di malignità della lesione purchè, in quest’ultimo caso, sia effettuato un immediato ed adeguato intervento laparotomico con scrupolosa toilette addominale. In ogni caso è opportuno limitare sempre lo spillage praticando l’aspirazione o biopsia all’interno del sacchetto endoscopico e praticando lavaggio ed aspirazione continua del punto di puntura o biopsia e di tutta la cavità peritoneale.
più recenti studi oncologici hanno dimostrato che la biopsia ovarica non fa peggiorare la prognosi anche in caso di malignità della lesione purchè, in quest’ultimo caso, sia effettuato un immediato ed adeguato intervento laparotomico con scrupolosa toilette addominale. In ogni caso è opportuno limitare sempre lo spillage praticando l’aspirazione o biopsia all’interno del sacchetto endoscopico e praticando lavaggio ed aspirazione continua del punto di puntura o biopsia e di tutta la cavità peritoneale.
Aspirazione del liquido peritoneale e washing: è la prima operazione da effettuare in caso di LPS o laparotomia. Qualora non si riesca ad individuare una sacca di liquido, si introduce in cavità 1 litro di soluzione fisiologica tiepida; la paziente viene fatta ruotare prima su un fianco e poi sull’altro in modo che il liquido si muova all’interno della cavità addominale; successivamente il liquido viene aspirato e si invia per l’esame citologico.
Ispezione lps accurata dell’ovaio in esame e di quello controlaterale, peritoneo, diaframma, omento, docce paracoliche, fegato ed intestino.
Thin-prep: solo in rari casi può offrire uno spunto diagnostico

ANATOMIA PATOLOGICA: Approssimativamente il 90% dei carcinomi ovarici sono tumori epiteliali che insorgono dall’epitelio ovarico di superficie o più probabilmente da cisti  di inclusione epiteliali. Alcuni studi hanno suggerito una possibile derivazione dai sistemi Mulleriani che comprendono cisti paraovariche e paratubariche, endometrio, endosalpingi ed epitelio della vescica urinaria[1]. Tutte queste porzioni del tratto genitale femminile riconoscono nell’epitelio celomatico (mesotelio) il precursore embriologico comune. La classificazione dei tumori epiteliali ovarici correntemente in uso, basata sulla morfologia delle cellule tumorali, li suddivide in: sierosi (60-70%), endometrioidi (10-20%), mucinosi (5-20%), a cellule chiare (3-10%)e indifferenziati.
di inclusione epiteliali. Alcuni studi hanno suggerito una possibile derivazione dai sistemi Mulleriani che comprendono cisti paraovariche e paratubariche, endometrio, endosalpingi ed epitelio della vescica urinaria[1]. Tutte queste porzioni del tratto genitale femminile riconoscono nell’epitelio celomatico (mesotelio) il precursore embriologico comune. La classificazione dei tumori epiteliali ovarici correntemente in uso, basata sulla morfologia delle cellule tumorali, li suddivide in: sierosi (60-70%), endometrioidi (10-20%), mucinosi (5-20%), a cellule chiare (3-10%)e indifferenziati.
Il carcinoma ovarico sieroso rappresenta l’istotipo più comune di carcinoma ovarico epiteliale. Caratterizzato da cellule che per morfologia e pattern di crescita ricordano l’epitelio tubarico. Istologicamente contrassegnato da invasione dello stroma, aree di crescita solida, papille complesse e possibili calcificazioni (i corpi psammomatosi). I carcinomi sierosi sono classificati in tumori sierosi di basso grado e tumori sierosi di alto grado sulla base dell’estensione delle atipie nucleari e delle mitosi. Morfologicamente i carcinomi sierosi di basso grado si caratterizzano per atipie nucleari minime e numero di mitosi basso, al contrario in quelli di alto grado si repertano atipie nucleari marcate ed elevato numero di mitosi. La dicotomia è conservata anche per il pattern genetico e di progressione.
Carcinoma ovarico endometrioide rappresenta il 10-20% di tutti i carcinomi ovarici epiteliali, con un’età media di insorgenza di 59 anni ed è istologicamente simile ai tumori maligni primitivi dell’endometrio. Nei soggetti affetti da carcinoma endometrioide sono stati occasionalmente riscontranti foci endometriosici in sede ovarica omo o controlaterale e/o extraovarica, e nel 14% dei casi un carcinoma endometrioide del corpo dell’utero. Macroscopicamente, il carcinoma endometrioide ha un aspetto solido-cistico (con aree cistiche contenenti materiale friabile e raccolte fluide), o, più raramente, totalmente solido con estese aree di necrosi ed emorragia. Microscopicamente è invece caratterizzato da spazi ghiandolari con proiezioni papillari rivestite da epitelio con stratificazione nucleare. Relativamente poco conosciuti i meccanismi molecolari di sviluppo del carcinoma endometrioide. Sembrano coinvolte mutazioni del gene della beta-catenina implicata nell’adesione cellulare e nella trasduzione del segnale. Disregolazioni del complesso catenina/caderina sono implicate nello sviluppo, progressione, invasione e metastasi di molte neoplasie[10,11]. Mutazioni del gene PTEN sono state ritrovate nel 43% delle donne con carcinoma ovarico endometrioide.
Carcinoma ovarico mucinoso Istologicamente i carcinomi mucinosi sono tumori costituiti da ghiandole, cisti o papille rivestite da cellule che contengono mucina (“globet cells” nel tipo intestinale); morfologicamente ricordano l’epitelio dell’endocervice e dell’intestino. Il carcinoma mucinoso si caratterizza per la presenza di atipie citologiche molto marcate: elevato numero di mitosi, nuclei ipercromatici, stratificazioni cellulari, ghiandole che infiltrano lo stroma e crescita solida. I tumori mucinosi sono spesso eterogenei, in particolare il tipo intestinale, e mostrano frequente coesistenza di elementi benigni, borderline e maligni nella singola neoplasia, suggerendo una chiara progressione della carcinogenesi da cistoadenoma a carcinoma invasivo (tumore borderline-tumore non invasivo-tumore microinvasivo e carcinoma invasivo). I meccanismi molecolari alla base della tumorigenesi, seppur ancora per lo più sconosciuti, riconoscono mutazioni di KRAS come evento precoce. I carcinomi mucinosi sono i tumori ovarici che raggiungono macroscopicamente le maggiori dimensioni seguiti dal tipo endometrioide (12).
Carcinoma ovarico a cellule chiare sono Tumori microscopicamente caratterizzati da grandi cellule con citoplasma chiaro contenente glicogeno che formano masse solide o strutture ghiandolari. Morfologicamente simile all’endometrio ipersecretivo, tanto da essere considerato una possibile variante del carcinoma endometrioide ovarico, colpisce in età più avanzata, è di solito associato ad una cattiva prognosi per la scarsa responsività alla chemioterapia. Nel 5-10% dei casi ci può essere un’associazione con foci endometriosici. Fra i meccanismi molecolari della carcinogenesi si segnala l’assenza di mutazioni del gene p53 come meccanismo anti-apoptotico tumorale e la presenza di metilazioni aberranti di TMS-1/ASC che funzionano fisiologicamente da silenziatori della trascrizione, mutazioni PTEN e disregolazioni di CD4461, una glicoproteina di membrana presente nelle cellule peritoneali che fa da recettore per GAG e acido ialuronico[13,14]. Sono stati inoltre osservati elevati livelli di instabilità dei microsatelliti (hMLH1 e hMSH2)euna bassa espressione di BAX nei pazienti con tumori chemioresitenti. Una comparazione del profilo genetico appartenente agli istotipi sieroso, endometrioideea cellule chiare dei carcinomi ovarici epiteliali dimostra 43 geni comuni a tutti gli istotipi suggerendo un processo di trasformazione maligna comune (11).

STADIAZIONE: Chiare indicazioni di stadiazione istologica e prognosi, usando strumenti sensibili quali la stadiazione FIGO e TNM sono essenziali. Indagini ecografiche e radiologiche addominali insieme alla laparoscopia consentono di ottenere una stadiazione della malattia. Si devono valutare la presenza e la quantità del liquido ascitico che viene inviato al laboratorio per l’esame citologico. Le ovaie vengono  esaminate per valutare la presenza di escrescenze, aderenze dense ‘ e rottura. E’ necessaria un’attenta ispezione del diaframma, della superficie peritoneale ed omentale sulle quali è opportuno effettuare un prelievo bioptico. Oltre all’isterectomia totale per via addominale e alla salpingo-ooforectomia bilaterale, è necessario procedere a un’omentectomia parziale e devono essere ispezionate le logge paracoliche. I linfonodi pelvici, così come quelli paraaortici nella regione dell’ilo renale, devono essere biopsiati. Dato che l’intervento chirurgico definisce lo stadio, stabilisce la prognosi e fornisce le indicazioni per la terapia successiva, deve essere eseguito da un chirurgo con competenza specifica nella stadiazione del cancro ovarico.
esaminate per valutare la presenza di escrescenze, aderenze dense ‘ e rottura. E’ necessaria un’attenta ispezione del diaframma, della superficie peritoneale ed omentale sulle quali è opportuno effettuare un prelievo bioptico. Oltre all’isterectomia totale per via addominale e alla salpingo-ooforectomia bilaterale, è necessario procedere a un’omentectomia parziale e devono essere ispezionate le logge paracoliche. I linfonodi pelvici, così come quelli paraaortici nella regione dell’ilo renale, devono essere biopsiati. Dato che l’intervento chirurgico definisce lo stadio, stabilisce la prognosi e fornisce le indicazioni per la terapia successiva, deve essere eseguito da un chirurgo con competenza specifica nella stadiazione del cancro ovarico.
PROGNOSI: La prognosi del cancro dell’ovaio dipende non solo dallo stadio, ma anche dall’estensione della malattia residua e dal grado istologico. Le pazienti che si presentano con malattia in fase avanzata, ma che risultano prive di residui significativi di malattia dopo la chirurgia, hanno una media di sopravvivenza di 39 mesi rispetto ai 17 mesi delle pazienti con resezione della massa tumorale subottimale. La prognosi nei tumori epiteliali dipende notevolmente anche dal grado istologico e in misura minore dall’istotipo. Alcuni studi condotti su pazienti con malattia in fase precoce hanno associato una sopravvivenza più lunga agli adenocarcinomi mucinosi piuttosto che agli istotipi endometrioide o sieroso e una prognosi peggiore ai carcinomi a cellule chiare. Le donne affette da neoplasia epiteliale maligna dell’ovaio allo stadio FIGO IA e IB e istologia favorevole (istotipi non a cellule chiare e ben differenziati) non sembrano beneficiare di alcuna terapia adiuvante postchirurgica. In tale gruppo di pazienti, infatti, la sopravvivenza libera da malattia è così alta dopo sola chirurgia (>90% a 6 anni) da non rendere necessaria l’adozione di un trattamento adiuvante.Donne affette da neoplasie allo stadio FIGO IA e IB e istologia “sfavorevole” (istotipi scarsamente differenziati o a cellule chiare), stadio Ic e stadio II, beneficiano di un trattamento chemioterapico adiuvante contenente platino.
Benché gli anatomopatologi utilizzino sistemi di grading diversi, in ogni sistema di grading la prognosi migliore è riservata ai tumori bene o moderatamente differenziati e quella peggiore ai sottotipi istologici scarsamente differenziati. La sopravvivenza tipica a 5 anni, indipendentemente dallo stadio della malattia, risulta essere la seguente: carcinoma ben differenziato, 88%; carcinoma moderatamente differenziato, 58%; carcinoma scarsamente differenziato, 27% (6).
 programma organizzato di screening nella popolazione femminile generale. Nella popolazione a rischio la ricerca di una familiarità positiva sta assumendo importanza clinica, alla luce di nuove conoscenze. Una familiarità positiva per carcinoma ovarico in due o più parenti di primo grado e la presenza di mutazioni nei geni responsabili della sindrome del carcinoma ereditario della mammella/ovaio (HBOC) e del carcinoma colon-rettale non polipoide del retto (HNPCC)si associano a un alto rischio di sviluppare il carcinoma ovarico.Nonostante esista una notevole varietà di parametri clinici, di laboratorio e di diagnostica
programma organizzato di screening nella popolazione femminile generale. Nella popolazione a rischio la ricerca di una familiarità positiva sta assumendo importanza clinica, alla luce di nuove conoscenze. Una familiarità positiva per carcinoma ovarico in due o più parenti di primo grado e la presenza di mutazioni nei geni responsabili della sindrome del carcinoma ereditario della mammella/ovaio (HBOC) e del carcinoma colon-rettale non polipoide del retto (HNPCC)si associano a un alto rischio di sviluppare il carcinoma ovarico.Nonostante esista una notevole varietà di parametri clinici, di laboratorio e di diagnostica
- Bibliografia:
- Il Cancro in Italia. I dati di incidenza dei Registri Tumori. Volume terzo: 1993-98. Plaxe SC. Epidemiology of low-grade serous ovarian cancer. Am J Obstet Gynecol 2008; 198: 459.e1-459.e9
- Vaidya AP et al. The follow-up of ovarian cancer. Sem Oncol 2003; 30: 401-12
- Jekmal A, Siegel R, Ward E, et al.: Cancer statistics, 2008. CA Cancer J Clin2008, 58:71–96
- Greenlee RT, Murray T, Bolden S, Wingo PA. 2000. Cancer statistics, 2000. CA Cancer J Clin 50: 7-33
- Kliewer EV, Smith KR. 1995. Ovarian cancer mortality among immigrants in Australia and Canada. Cancer Epidemiol Biomarkers Prev 4: 453-458
- Singer G, Oldt R, 3rd, Cohen Y, et al: Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst 2003; 95:484-6
- Mirjam J.A. Engelen, M.D.Henk W.A. de Bruijn, Ph.D. et al: “Serum CA 125, Carcinoembryonic Antigen, and CA 19-9 as Tumor Markers in Borderline Ovarian Tumors”.Gynecologic Oncology July 2000 Volume 78, Issue 1, Pages 16–20
- Berrino F et al (eds). Survival of cancer patients in Europe: the EUROCARE-2 Study. IARC ScientificPublications No 151. Lyon: IARC Press, 1999
- Bell R et al. Screening for ovarian cancer: a systematic review. Health Technol Assess 1998; 2:1-84.
- Jacobs IJ et al. Screening for ovarian cancer: a pilot randomised controlled trial. Lancet 1999; 353:1207-10.
- Einhorn N et al. Long-term follow-up of the Stockholm screening study on ovarian cancer. Gynecol Oncol 2000; 79: 466-70.
- Brunet JS et al. Effect of smoking on breast cancer in carriers of mutant BRCA1 and BRCA2 genes. J Natl Cancer Inst 1998; 90; 761-66.
- Narod SA et al. Oral contraceptives and the risk of hereditary ovarian cancer. New Engl J Med 1998;339: 424-28.
- Narod SA et al. Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations: a case control study. Lancet 2001; 357: 1467-70
- Smith LH et al. Detection of malignant ovarian neoplasm(s): a review of the literature. I. Detection of the patient at risk; clinical, radiological and cytological detection. Obstet Gynecol Surv 1984; 39:313-28.
- Yancik R et al. Ovarian cancer in the elderly: an analysis of surveillance, epidemiology, and end results program data. Am. J. Obstet. Gynecol. 1986; 154: 639-47.Easton DF et al. Cancer mortality in relatives of women with ovarian cancer: the OPCS study. Int JCancer 1996; 65: 284-94
- Sutcliffe S et al. Ovarian cancer risks to women in families with two or more cases of ovarian cancer. Int J Cancer 2000; 87: 110-17.Wheeler JM et al. DNA mismatch repair genes and colorectal cancer. Gut 2000; 47:148-53.
- Aarnio M et al. Cancer risk in mutation carriers of DNA-mismatch repair genes. Int J Cancer 1999;81: 214-18
- Coughlin SS et al. A meta-analysis of estrogen replacement therapy and risk of epithelial ovarian cancer. J Clin Epidemiol 2000; 53: 367-75.
- Lacey JV et al. Menopausal hormone replacement therapy and risk of ovarian cancer. JAMA 2002;288: 334-41.
- Hankinson SE et al. A quantitative assessment of oral contraceptive use and risk of ovarian cancer. Obstet Gynecol 1992; 80: 708-14.
- The reduction in risk of ovarian cancer associated with oral-contraceptive use. The Cancer and steroid Hormone Study of the Centers for Disease Control and the National Institute of Child Health and Human Development. N Engl J Med 1987; 316: 650-55.
- Piver MS et al. Familial ovarian cancer. A report of 658 families from the Gilda Radner Familial Ovarian Cancer Registry 1981-1991. Cancer 1993; 71: 582-88.
- Modan B et al. Parity, oral contraceptives, and the risk of ovarian cancer among carriers and non-carriers of a BRCA1 or BRCA2 Mutation. N Engl J Med 2001; 345: 235-40.
- Ursin G et al. Does oral contraceptive use increase the risk of breast cancer in women with BRCA1/BRCA2 mutations more than in other women? Cancer Res 1997; 57: 3678–81.
- Kennedy RD et al. BRCA1: mechanisms of inactivation and implications for management of patients. Lancet 2002; 360: 1007-14
- National Cancer Institute. Ovarian Cancer (PDQ®) Prevention http://www.cancer.gov/cancerinfo/pdq/prevention/ovarian/)
- Hankinson SE et al. A prospective study of reproductive factors and risk of epithelial ovarian can-cer. Cancer 1995; 76: 284-90
- Narod SA et al. Risk modifiers in carriers of BRCA1 mutations. Int J Cancer 1995; 64: 394-98
- Godard B et al. Risk factors for familial and sporadic ovarian cancer among French Canadians: acase-control study. Am J Obstet Gynecol 1998; 179: 403-10
- Tobacman JK et al. Intra-abdominal carcinomatosis after prophylactic oophorectomy in ovarian-cancer-prone families. Lancet 1982; 2: 795-97.
- Piver MS et al. Primary peritoneal carcinoma after prophylactic oophorectomy in women with a family history of ovarian cancer. A report of the Gilda Radner Familial Ovarian Cancer Registry. Cancer 1993; 71: 2751-55.
- Struewing JP et al. Prophylactic oophorectomy in inherited breast/ovarian cancer families. J Natl Cancer Inst Monogr 1995; 33-35.Hankinson SE et al. Tubal ligation, hysterectomy, and risk of ovarian cancer. A prospective study.JAMA 1993; 270: 2813-18
- Narod SA et al. Tubal ligation and risk of ovarian cancer in carriers of BRCA1 or BRCA2 mutations:a case control study. Lancet 2001; 357: 1467-70
- Farrow DC, Weiss NS, Lyon JL, Daling JR: Association of obesity and ovarian cancer in a case-control study. American Journal of Epidemiology [1989, 129(6):1300-1304]
- Catherine M. Olsen et al: “Obesity and the risk of epithelial ovarian cancer: A systematic review and meta-analysis”. European Journal of Cancer Volume 43, Issue 4, March 2007, Pages 690–709
- Kathy J. Helzlsouer et al: “Women with low serum gonadotropin levels or high androgen levels have an increased risk of ovarian cancer”. JAMA. 1995;274(24):1926-1930
- Jayson GC, Kohn EC, Kitchener HC, Ledermann JA (October 2014). “Ovarian cancer”. Lancet384 (9951): 1376–88.
- Manson, JoAnn E.; Bassuk, Shari S. (2012). “The Menopause Transition and Postmenopausal Hormone Therapy”. In Longo DL, Kasper DL, Jameson JL, Fauci AS, Hauser SL, Loscalzo J. Harrison’s Principles of Internal Medicine (18th ed.). McGraw-Hill
- MCCluggage WG. Morphological subtypes of ovarian carcinoma: a review with emphasis on new developments and pathogenesis. Pathology 2011;43: 420-32.
- Lalwani N, Shanbhogue AK, Vikram R, et al. Current update on borderline ovarian neoplasm. AJR Am J Roentgenol 2010;194:330-6.
- Fathalla MF. Incessant ovulation – a factor in ovarian neoplasia? Lancet 1971;2:163.
- Gambacciani M, Monteleone P et al. Hormone replacement therapy andendometrial, ovarian and colorectal cancer,Bets Pract Res Clin Endocrinol Metab 2003;17 (1): 139-47.
- Miracle-McMahill HL, Calle EE, Kosinski AS, et al. Tubal ligation and fatal ovarian cancer in a large prospective cohort study. Am J Epidemiol 1997;145:349-57.
- Brinton LA, Gridley G, Persson I, et al. Cancer risk after a hospital discharge diagnosis of endometriosis. Am J Obstet Gynecol 1997;176:572-9.
- Borgfeldt C, Andolf E. Cancer risk after hospital discharge diagnosis of benignovarian cysts and endometriosis. Acta Obstet Gynecol Scand 2004;(83):395-400.
- Gnagy S, Ming EE, Devesa SS, et al. Declining ovarian cancer rates in US women in relation to parity and oral contraceptive use. Epidemiology 2000;11:102-5.
- Jordan SJ , Whiteman DC, Purdie DM, et al. Does smoking increase risk of ovarian cancer? A systematic review. Gynecol Oncol. 2006; 103(3):1122-9. Epub 2006 Sep 2.
- Harlow BL, Cramer DW, Bell DA, Welch WR. Perineal exposure to talc and ovarian cancer risk. Obstet Gynecol. 1992; 80(1):19-26.
- Huncharek M, Muscat J, Onitilo A, Kupelnick B. Use of cosmetic talc on contraceptive diaphragms and risk of ovarian cancer: a meta-analysis of nine observational studies. Eur J Cancer Prev. 2007;16(5):422-429.
- Sherman ME. Cytopathology. In: Kurman RJ (ed) Blaunstein’s pathology of the female genital tract. New York: Springer, 1994 (4th ed), 1097-130.
- Bibbo M et al. Peritoneal washings and ovary. In: Bibbo M (ed) Comprehensive cytopathology. Philadelphia: Saunders, 1997 (2nd ed), 315-23.
- Naylor B. Pleural, peritoneal and pericardial fluids. In Bibbo M. (ed) Comprehensive cytopathology. Philadelphia: Saunders, 1997 (2nd ed), 551-621.
- Zuna RE et al. Peritoneal washing cytology in gynecologic cancers: long-term follow-up of 355 patients. J Natl Cancer Inst 1996; 88: 980-87.Griffiths CT. Surgical resection of tumor bulk in the primary treatment of ovarian carcinoma. NatlCancer Inst Monogr 1975; 42: 101-04
- Tidy J et al. Endometrioid carcinoma of the ovary: a retrospective study. Br J Obstet Gynaecol 1988;95: 1165-69
- Griffiths CT. Surgical resection of tumor bulk in the primary treatment of ovarian carcinoma. Natl Cancer Inst Monogr 1975; 42: 101-04.
- Hoskins WJ et al. The influence of cytoreductive surgery on recurrent-free interval and survival in small-volume stage III epithelial ovarian cancer: a Gynecologic Oncology Group study. Gynecol Oncol 1992; 47: 159-66.
- Omura GA et al. Long follow-up and prognostic factor analysis in advanced ovarian carcinoma: the Gynecologic Oncology Group experience. J Clin Oncol 1991; 9: 1138-50.
- van der Burg ME et al. The effect of debulking surgery after induction chemotherapy on the prognosis in advanced epithelial ovarian cancer. Gynecological Cancer Cooperative Group of the European Organization for Research and Treatment of Cancer. N Engl J Med 1995; 332: 629-34.
- Vergote I et al. Neoadjuvant chemotherapy or primary debulking surgery in advanced ovarian carcinoma: a retrospective analysis of 285 patients. Gynecol Oncol 1998; 71: 431-36
- Young RC et al. Adjuvant therapy in stage I and stage II epithelial ovarian cancer. N. Engl J Med1990; 322: 1021-27
- Advanced Ovarian Cancer Trialist Group: Chemotherapy in advanced ovarian cancer: an overview of randomised clinical trials. Br Med J 1991 303: 884-93.
-
Advanced Ovarian Cancer Trialist Group. Chemotherapy in advanced ovarian cancer: four systematic mata-analyses of individual patient data from 37 randomised trials. Advanced Ovarian Cancer Trialist Group. Br J Cancer 1998; 78: 1479-87.
-
Ovarian Cancer Meta-analysis Project. Cyclophosphamide plus cisplatin versus cyclophosphamide, doxorubicin and cisplatin chemotherapy of ovarian carcinoma: a meta-analysis. J Clin Oncol 1991; 9: 1668-74.
-
A’Hern RP et al. Impact of doxorubicin on survival in advanced ovarian cancer. J Clin Oncol 1995;13: 726-32.
- Mc Guire WP et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patient with stage III and stage IV ovarian cancer. N Engl J Med 1996; 334: 1-6.
- Piccart MJ et al. Randomised intergroup trial of cisplatin-paclitaxel versus cisplatin-cyclophosphamide in women with advanced epithelial ovarian cancer: three-year result. J Natl Cancer I 2000;92: 699-708.
-
Muggia FM et al. Phase III randomized study of cisplatin versus paclitaxel versus cisplatin and paclitaxel in patients with suboptimal stage III or IV ovarian cancer: A gynecologic oncology group study. J Clin Oncol 2000; 18: 106-15.
-
The ICON collaborators. Paclitaxel plus carboplatin versus standard chemotherapy with either single-agent carboplatin or cyclophosphamide, doxorubicin, and cisplatin in women with ovarian cancer: the ICON 3 randomized trials. Lancet 2002; 360: 505-15.
-
Du Bois A et al. Cisplatin/paclitaxel versus carboplatin/paclitaxel in ovarian cancer: update of an Arbeitsgemeinschaft Gynaekologische Onkologie (AGO) Study Group trial. Proc Am Soc Clin Oncol 1999; 18: 356a.
-
Ozols RF et al. Randomized phase III study of cisplatin (CIS)/paclitaxel (PAC) versus carboplatin (carbo)/PAC in optimal stage III epithelial ovarian cancer (OC): a Gynecologic Oncology Group trial (GOG 158). Proc Am Soc Clin Oncol 1999; 18: 356a.
-
Neijt JP et al. Exploratory phase III study of paclitaxel and cisplatin versus paclitaxel and carboplatin in advanced ovarian cancer. J Clin Oncol 2000; 18: 3084-92.
- Vaidya AP et al. The follow-up of ovarian cancer. Sem Oncol 2003; 30: 401-12.
- Rustin GJS et al. Tumor markers. Ann Oncol 1993; 4(Suppl4): 71-77.
- Markman M. Follow-up of the asyntomatic patient with ovarian cancer. Gynecol Oncol 1994; 55:S134-37.
-
Prayer L et al. CT and MR accurancy in the detection of tumor recurrence in patients treated for ovarian cancer. J Comput Assist Tomogr 1993; 17: 626-32.
- Salmon E et al. Management of recurrent ovarian cancer: evidence- based decision. Curr Opin Oncol 2002; 14: 519-27.
- Markman M et al. Second-line platinum therapy in patients with ovarian cancer previously trea-ted with cisplatin. J Clin Oncol 1991; 9: 389-93
- The ICON and AGO Collaborators. Paclitaxel plus platinum-based chemotherapy versus conventional platinum-based chemotherapy in women with relapsed ovarian cancer: the ICON 4/AGO_OVAR-2.2 trial. Lancet 2003; 1361: 2099-106.
- Donahue VC et al. Sexual rehabilitation of gynecologic cancer patients. Obstet Gynecol 1977; 49:118-21.
-
Andersen BL et al. Sexual functioning among breast cancer, gynecologic cancer and healthy women. J Counsel Clin Psychol 1985; 53: 25-32.
-
Auchinloss SS. Sexual dysfunction in cancer patients: issues in evaluation and treatment. In: Holland JC and Rowland JH (eds) Handbook of Psycho-Oncology., New York: Oxford University Press, 1989
- Boyd J, Rubin SC. 1997. Hereditary ovarian cancer: molecular genetics and clinical implications [Review]. Gynecol Oncol 64: 196-206
- Goldberg JM, Piver MS, Jishi MF, Blumenson L. 1997. Age at onset of ovarian cancer in women with a strong family history of ovarian cancer. Gynecol Oncol 66:3-9
- Lynch HT, Smyrk T. 1996. Hereditary nonpolyposis colorectal cancer (Lynch syndrome). An updated review [Review]. Cancer 78: 1149-1167
- R Wooster, SL Neuhausen, J Mangion et al: “Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13”. Science 1994;265,5181:2088-2090
- Paley J.P. et al: Occult Cancer of the Fallopian Tube in BRCA-1 Germline Mutation Carriers at Prophylactic Oophorectomy: A Case for Recommending Hysterectomy at Surgical Prophylaxis.
Gynecologic Oncology Volume 80, Issue 2, February 2001, Pages 176–180
- Nicoletto M.O. et al: BRCA-1 and BRCA-2 mutations as prognostic factors in clinical practice and genetic counselling. Cancer Treatment Reviews. Volume 27, Issue 5, October 2001, Pages 295–304
- Lancaster JM et al: BRCA2 mutation in primary breast and ovarian cancer. Nature genetic 1996,13.
-
Lehrer S. et al: Absence of 185delAG mutation of the BRCA1 gene and 6174delT mutation of the BRCA2 gene in Ashkenazi Jewish men with prostate cancer. Br J Cancer. 1998 Sep; 78(6): 771–773.
-
Boring CC, Squires TS, Tong T, Montgomery S. Cancer statistics, 1994. CA Cancer J Clin. 1994 Jan-Feb;44(1):7–26.
-
Eastman P. Search for prostate cancer gene sites may succeed in 1996. J Natl Cancer Inst. 1996 Jul 17;88(14):952–953.
-
FitzGerald MG, MacDonald DJ, Krainer M, Hoover I, O’Neil E, Unsal H, Silva-Arrieto S, Finkelstein DM, Beer-Romero P, Englert C, et al. Germ-line BRCA1 mutations in Jewish and non-Jewish women with early-onset breast cancer. N Engl J Med. 1996 Jan 18;334(3):143–149.
-
Ford D, Easton DF, Bishop DT, Narod SA, Goldgar DE. Risks of cancer in BRCA1-mutation carriers. Breast Cancer Linkage Consortium. Lancet. 1994 Mar 19;343(8899):692–695.
-
Friedman LS, Szabo CI, Ostermeyer EA, Dowd P, Butler L, Park T, Lee MK, Goode EL, Rowell SE, King MC. Novel inherited mutations and variable expressivity of BRCA1 alleles, including the founder mutation 185delAG in Ashkenazi Jewish families. Am J Hum Genet. 1995 Dec;57(6):1284–1297.
-
Gao X, Zacharek A, Grignon DJ, Sakr W, Powell IJ, Porter AT, Honn KV. Localization of potential tumor suppressor loci to a < 2 Mb region on chromosome 17q in human prostate cancer. Oncogene. 1995 Oct 5;11(7):1241–1247. [PubMed]
-
Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, Honn KV. Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 1995 Mar 1;55(5):1002–1005. [PubMed]
-
Giovannucci E, Tosteson TD, Speizer FE, Ascherio A, Vessey MP, Colditz GA. A retrospective cohort study of vasectomy and prostate cancer in US men. JAMA. 1993 Feb 17;269(7):878–882. [PubMed
-
Isaacs SD, Kiemeney LA, Baffoe-Bonnie A, Beaty TH, Walsh PC. Risk of cancer in relatives of prostate cancer probands. J Natl Cancer Inst. 1995 Jul 5;87(13):991–996.
-
Langston AA, Malone KE, Thompson JD, Daling JR, Ostrander EA. BRCA1 mutations in a population-based sample of young women with breast cancer. N Engl J Med. 1996 Jan 18;334(3):137–142.
-
Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994 Oct 7;266(5182):66–71. [PubMed]
-
Oddoux C, Struewing JP, Clayton CM, Neuhausen S, Brody LC, Kaback M, Haas B, Norton L, Borgen P, Jhanwar S, et al. The carrier frequency of the BRCA2 6174delT mutation among Ashkenazi Jewish individuals is approximately 1%. Nat Genet. 1996 Oct;14(2):188–190. [PubMed]
-
Pienta KJ, Esper PS. Risk factors for prostate cancer. Ann Intern Med. 1993 May 15;118(10):793–803.
-
Sellers TA, Potter JD, Rich SS, Drinkard CR, Bostick RM, Kushi LH, Zheng W, Folsom AR. Familial clustering of breast and prostate cancers and risk of postmenopausal breast cancer. J Natl Cancer Inst. 1994 Dec 21;86(24):1860–1865. [PubMed]
-
Smith JR, Freije D, Carpten JD, Grönberg H, Xu J, Isaacs SD, Brownstein MJ, Bova GS, Guo H, Bujnovszky P, et al. Major susceptibility locus for prostate cancer on chromosome 1 suggested by a genome-wide search. Science. 1996 Nov 22;274(5291):1371–1374.
-
Stephenson J. Prostate cancer gene hunters track their quarry. JAMA. 1996 Sep 18;276(11):861–863. [PubMed]
-
Struewing JP, Abeliovich D, Peretz T, Avishai N, Kaback MM, Collins FS, Brody LC. The carrier frequency of the BRCA1 185delAG mutation is approximately 1 percent in Ashkenazi Jewish individuals. Nat Genet. 1995 Oct;11(2):198–200. [PubMed]
-
Struewing JP, Hartge P, Wacholder S, Baker SM, Berlin M, McAdams M, Timmerman MM, Brody LC, Tucker MA. The risk of cancer associated with specific mutations of BRCA1 and BRCA2 among Ashkenazi Jews. N Engl J Med. 1997 May 15;336(20):1401–1408.
-
Whittemore AS, Wu AH, Kolonel LN, John EM, Gallagher RP, Howe GR, West DW, Teh CZ, Stamey T. Family history and prostate cancer risk in black, white, and Asian men in the United States and Canada. Am J Epidemiol. 1995 Apr 15;141(8):732–740.
-
Williams BJ, Jones E, Zhu XL, Steele MR, Stephenson RA, Rohr LR, Brothman AR. Evidence for a tumor suppressor gene distal to BRCA1 in prostate cancer. J Urol. 1996 Feb;155(2):720–725.
-
Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D, et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13. Science. 1994 Sep 30;265(5181):2088–2090.
- Amy Finch et al: “Salpingo-oophorectomy and the Risk of Ovarian, Fallopian Tube, and Peritoneal Cancers in Women With a BRCA1 or BRCA2 Mutation”. JAMA. 2006;296(2):185-192. doi:10.1001/jama.296.2.185
- Tobacman JK, Greene MH, Tucker MA. et al. Intra-abdominal carcinomatosis after prophylactic oophorectomy in ovarian-cancer-prone families. Lancet. 1982;2:795-797
- Piver MS, Jishi MF, Tsukada Y, Nava G. Primary peritoneal carcinoma after prophylactic oophorectomy in women with a family history of ovarian cancer: a report of the Gilda Radner Familial Ovarian Cancer Registry. Cancer. 1993;71:2751-2755
- Rebbeck TR, Lynch HT, Neuhausen SL. et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2 mutations. N Engl J Med. 2002;346:1616-1622
- Olivier RI, van Beurden M, Lubsen MA. et al. Clinical outcome of prophylactic oophorectomy in BRCA1/BRCA2 mutation carriers and events during follow-up. Br J Cancer. 2004;90:1492-1497
- Rutter JL, Wacholder S, Chetrit A. et al. Gynecologic surgeries and risk of ovarian cancer in women with BRCA1 and BRCA2 Ashkenazi founder mutations: an Israeli population-based case-control study. J Natl Cancer Inst. 2003;95:1072-1078
- Kauff ND, Satagopan JM, Robson ME. et al. Risk-reducing salpingo-oophorectomy in women with a BRCA1 or BRCA2 mutation. N Engl J Med. 2002;346:1609-1615
- Powell BC, Kenley E, Chen L. et al. Risk-reducing salpingo-oophorectomy in BRCA mutation carriers: role of serial sectioning in the detection of occult malignancy. J Clin Oncol. 2005;23:127-132
- Chen KT, Schooley JL, Flam MS. Peritoneal carcinomatosis after prophylactic oophorectomy in familial ovarian cancer syndrome. Obstet Gynecol. 1985;66:(suppl 3) 93S-94S
- Paley PJ, Swisher EM, Garcia RL. et al. Occult cancer of the fallopian tube in BRCA1 germline mutation carriers at prophylactic oophorectomy: a case for recommending hysterectomy at surgical prophylaxis. Gynecol Oncol. 2001;80:176-180
- Colgan TJ, Boerner SL, Murphy KJ, Cole DE, Narod SA, Rosen B. Peritoneal lavage cytology: an assessment of its value during prophylactic oophorectomy. Gynecol Oncol. 2002;85:397-403
- Finch A, Shaw P, Rosen B, Murphy J, Narod SA, Colgan TJ. Clinical and pathologic findings of prophylactic salpingo-oophorectomies in 159 BRCA1 and BRCA2 carriers [epub]. Gynecol Oncol. 2005;100:58-64
- Easton DF. How many more breast cancer predisposition genes are there? Breast Cancer Research 1999; 1(1):14–17.
- Campeau PM, Foulkes WD, Tischkowitz MD. Hereditary breast cancer: New genetic developments, new therapeutic avenues. Human Genetics 2008; 124(1):31–42
- Pal T, Permuth-Wey J, Betts JA, et al. BRCA1 and BRCA2 mutations account for a large proportion of ovarian carcinoma cases. Cancer 2005; 104(12):2807–16
- Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 penetrance. Journal of Clinical Oncology 2007; 25(11):1329–1333
- Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. Journal of the National Cancer Institute 2007; 99(23):1811–1814
- Levy-Lahad E, Friedman E. Cancer risks among BRCA1 and BRCA2 mutation carriers. British Journal of Cancer 2007; 96(1):11–15
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP (October 1999). “Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association”. Cell 99 (3): 323–34.
- Steck PA, Pershouse MA, Jasser SA, Yung WK, Lin H, Ligon AH, Langford LA, Baumgard ML, Hattier T, Davis T, Frye C, Hu R, Swedlund B, Teng DH, Tavtigian SV (Apr 1997). “Identification of a candidate tumour suppressor gene, MMAC1, at chromosome 10q23.3 that is mutated in multiple advanced cancers”. Nature Genetics 15 (4): 356–62.
- “OrthoMaM phylogenetic marker: PTEN coding sequence”. “Entrez Gene: PTEN phosphatase and tensin homolog (mutated in multiple advanced cancers 1)”.
- Chu EC, Tarnawski AS (Oct 2004). “PTEN regulatory functions in tumor suppression and cell biology”. Medical Science Monitor 10 (10): RA235–41
- Shi Y, Wang J, Chandarlapaty S, Cross J, Thompson C, Rosen N, Jiang X (Jun 2014). “PTEN is a protein tyrosine phosphatase for IRS1”. Nature Structural & Molecular Biology 21 (6): 522–7.
- Shnitsar I, Bashkurov M, Masson GR, Ogunjimi AA, Mosessian S, Cabeza EA, Hirsch CL, Trcka D, Gish G, Jiao J, Wu H, Winklbauer R, Williams RL, Pelletier L, Wrana JL, Barrios-Rodiles M (2015-09-24). “PTEN regulates cilia through Dishevelled”. Nature Communications 6:388.
- Haynie DT, Xue B (Feb 2015). “Superdomains in the protein structure hierarchy: The case of PTP-C2”. Protein Science 24: 874–82.
- Campbell RB, Liu F, Ross AH (Sep 2003). “Allosteric activation of PTEN phosphatase by phosphatidylinositol 4,5-bisphosphate”. The Journal of Biological Chemistry 278 (36): 33617–20.
- Iijima M, Huang YE, Luo HR, Vazquez F, Devreotes PN (Apr 2004). “Novel mechanism of PTEN regulation by its phosphatidylinositol 4,5-bisphosphate binding motif is critical for chemotaxis”. The Journal of Biological Chemistry 279(16): 16606–13.
- McConnachie G, Pass I, Walker SM, Downes CP (May 2003). “Interfacial kinetic analysis of the tumour suppressor phosphatase, PTEN: evidence for activation by anionic phospholipids”. The Biochemical Journal 371 (Pt 3): 947–55.
- Rahdar M, Inoue T, Meyer T, Zhang J, Vazquez F, Devreotes PN (Jan 2009). “A phosphorylation-dependent intramolecular interaction regulates the membrane association and activity of the tumor suppressor PTEN”. Proceedings of the National Academy of Sciences of the United States of America 106 (2): 480–5.
- Masson GR, Perisic O, Burke JE, Williams RL (Nov 2015). “The intrinsically disordered tails of PTEN and PTEN-L have distinct roles in regulating substrate specificity and membrane activity”. The Biochemical Journal
- Hopkins BD, Fine B, Steinbach N, Dendy M, Rapp Z, Shaw J, Pappas K, Yu JS, Hodakoski C, Mense S, Klein J, Pegno S, Sulis ML, Goldstein H, Amendolara B, Lei L, Maurer M, Bruce J, Canoll P, Hibshoosh H, Parsons R (Jul 2013). “A secreted PTEN phosphatase that enters cells to alter signaling and survival”. Science 341 (6144): 399–402.
- Liang H, He S, Yang J, Jia X, Wang P, Chen X, Zhang Z, Zou X, McNutt MA, Shen WH, Yin Y (May 2014). “PTENα, a PTEN isoform translated through alternative initiation, regulates mitochondrial function and energy metabolism”.Cell Metabolism 19 (5): 836–48.
- Malaney P, Uversky VN, Davé V (Nov 2013). “The PTEN Long N-tail is intrinsically disordered: increased viability for PTEN therapy”. Molecular bioSystems 9 (11): 2877–88.
- Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP (Aug 2005). “Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis”. Nature 436 (7051): 725–30.
- Pilarski R, Eng C (May 2004). “Will the real Cowden syndrome please stand up (again)? Expanding mutational and clinical spectra of the PTEN hamartoma tumour syndrome”. Journal of Medical Genetics 41 (5): 323–6.
- Napoli E, Ross-Inta C, Wong S, Hung C, Fujisawa Y, Sakaguchi D, Angelastro J, Omanska-Klusek A, Schoenfeld R, Giulivi C (2012). “Mitochondrial dysfunction in Pten haplo-insufficient mice with social deficits and repetitive behavior: interplay between Pten and p53”. PLOS ONE 7 (8): e42504.
- “Rodent of the Week: Nerves regenerated after spinal cord injury”. The Los Angeles Times. August 13, 2010.
- Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z (Sep 2010).“PTEN deletion enhances the regenerative ability of adult corticospinal neurons”.Nature Neuroscience 13 (9): 1075–81. doi:10.1038/nn.2603. PMC 2928871.PMID 20694004.
- Walker, C. L.; Walker, M. J.; Liu, N. K.; Risberg, E. C.; Gao, X; Chen, J; Xu, X. M. (2012). “Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury”. PLoS ONE 7 (1): e30012.
- Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang SI, Puc J, Miliaresis C, Rodgers L, McCombie R, Bigner SH, Giovanella BC, Ittmann M, Tycko B, Hibshoosh H, Wigler MH, Parsons R (Mar 1997). “PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer”.Science 275 (5308): 1943–7.
- Miller SJ, Lou DY, Seldin DC, Lane WS, Neel BG (Sep 2002). “Direct identification of PTEN phosphorylation sites”. FEBS Letters 528 (1-3): 145–53.doi:10.1016/S0014-5793(02)03274-X. PMID 12297295.
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky LA (Jul 2000). “Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase”. The Journal of Biological Chemistry 275 (28): 21477–85.
- Yu Z, Fotouhi-Ardakani N, Wu L, Maoui M, Wang S, Banville D, Shen SH (Oct 2002). “PTEN associates with the vault particles in HeLa cells”. The Journal of Biological Chemistry 277 (43): 40247–52. doi:10.1074/jbc.M207608200.PMID 12177006.
- Wang X, Shi Y, Wang J, Huang G, Jiang X (Sep 2008). “Crucial role of the C-terminus of PTEN in antagonizing NEDD4-1-mediated PTEN ubiquitination and degradation”. The Biochemical Journal 414 (2): 221–9.doi:10.1042/BJ20080674. PMID 18498243.
- Lin HK, Hu YC, Lee DK, Chang C (Oct 2004). “Regulation of androgen receptor signaling by PTEN (phosphatase and tensin homolog deleted on chromosome 10) tumor suppressor through distinct mechanisms in prostate cancer cells”. Molecular Endocrinology 18 (10): 2409–23.
- Freeman DJ, Li AG, Wei G, Li HH, Kertesz N, Lesche R, Whale AD, Martinez-Diaz H, Rozengurt N, Cardiff RD, Liu X, Wu H (Feb 2003). “PTEN tumor suppressor regulates p53 protein levels and activity through phosphatase-dependent and -independent mechanisms”. Cancer Cell 3 (2): 117–30.
- Tamura M, Gu J, Danen EH, Takino T, Miyamoto S, Yamada KM (Jul 1999). “PTEN interactions with focal adhesion kinase and suppression of the extracellular matrix-dependent phosphatidylinositol 3-kinase/Akt cell survival pathway”. The Journal of Biological Chemistry 274 (29): 20693–703.
- Haier J, Nicolson GL (Feb 2002). “PTEN regulates tumor cell adhesion of colon carcinoma cells under dynamic conditions of fluid flow”. Oncogene 21 (9): 1450–60.
- Prat J, Ribé A, Gallardo A.; “Hereditary ovarian cancer”. Hum Pathol. 2005 Aug;36(8):861-70.
- [Genetic predisposition and ovarian cancer]. Coupier I, Gauthier-Villars M, This P, Stoppa-Lyonnet D. Rev Prat. 2004 Oct 31; 54(16):1757-62.
- Tomkinson H, Kemp J, Oliver S, Swaisland H, Taboada M, Morris T (2011). “Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: two open-label comparative studies”. BMC Clin Pharmacol 11: 3.
- Trump DL, Payne H, Miller K, et al. (September 2011). “Preliminary study of the specific endothelin a receptor antagonist zibotentan in combination with docetaxel in patients with metastatic castration-resistant prostate cancer”. Prostate 71 (12): 1264–75.
- Kimonis V, Goldstein A, Pastakia B, Yang M, Kase R, DiGiovanna J, Bale A, Bale S (1997). “Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome”. Am J Med Genet 69 (3): 299–308.
- Gorlin R, Goltz R (1960). “Multiple nevoid basal-cell epithelioma, jaw cysts and bifid rib. A syndrome”. N Engl J Med 262 (18): 908–12.
- Johnson R, Rothman A, Xie J, Goodrich L, Bare J, Bonifas J, Quinn A, Myers R, Cox D, Epstein E, Scott M (1996). “Human homolog of patched, a candidate gene for the basal cell nevus syndrome”. Science 272 (5268): 1668–71.
- Farrow DC, Weiss N, Lyon JL and Dalin JR: ” Association of obesity and ovarian cancer in a case-control study”. American Journal of Epidemiology [1989, 129(6):1300-1304]
- Olsen CM et al: “Obesity and the risk of epithelial ovarian cancer: A systematic review and meta-analysis”. European Journal of Cancer 2007;43,4:690-709.
- Fairfield KM et al: “Obesity, weight gain, and ovarian cancer”. Obstetrics & Gynecology 2002;100,2:288-296.
- Kathy J. Helzlsouer et al: “Serum Gonadotropins and Steroid Hormones and the Development of Ovarian Cancer”. JAMA. 1995;274(24):1926-1930
- Rodriguez C, Calle EE, Fakhrabadi-Shohoohi, Jacobs EJ and Thun MJ: “Body Mass Index, Height, and the Risk of Ovarian Cancer Mortality in a Prospective Cohort of Postmenopausal Women”. Cancer Epidemiol Biomarkers Prev September 2002 11; 822
- Annekatrin Lukanova and Rudolf Kaaks Endogenous Hormones and Ovarian Cancer: Epidemiology and Current Hypotheses. Cancer Epidemiol Biomarkers Prev January 2005 14; 98
- avid M., Christopher J. Bai , Penelope M., David C. Whiteman, Sandi Pirozzo, Adèle C. Green Body size and ovarian cancer: case–control study and systematic review (Australia) Cancer Causes & Control November 2001, Volume 12, Issue 9,pp 855-863
-
Kingsley M. Ekumi, Hana Paculova, Tina Lenasi, Vendula Pospichalova, Christian A. Bösken, Jana Rybarikova, Vitezslav Bryja,Matthias Geyer, Dalibor Blazek, and Matjaz Barboric: Ovarian carcinoma CDK12 mutations misregulate expression of DNA repair genes via deficient formation and function of the Cdk12/CycK complex. Nucleic Acids Res. 2015 Mar 11; 43(5): 2575–2589. -
Bartkowiak B., Liu P., Phatnani H.P., Fuda N.J., Cooper J.J., Price D.H., Adelman K., Lis J.T., Greenleaf A.L. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–2316. -
Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., Peterlin B.M. The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 2011;25:2158–2172. -
Cheng S.W., Kuzyk M.A., Moradian A., Ichu T.A., Chang V.C., Tien J.F., Vollett S.E., Griffith M., Marra M.A., Morin G.B. Interaction of cyclin-dependent kinase 12/CrkRS with cyclin K1 is required for the phosphorylation of the C-terminal domain of RNA polymerase II. Mol. Cell. Biol. 2012;32:4691–4704. -
Bosken C.A., Farnung L., Hintermair C., Merzel Schachter M., Vogel-Bachmayr K., Blazek D., Anand K., Fisher R.P., Eick D., Geyer M. The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 2014;5:3505. -
Joshi P.M., Sutor S.L., Huntoon C.J., Karnitz L.M. Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 2014;289:9247–9253. -
Chen H.H., Wang Y.C., Fann M.J. Identification and characterization of the CDK12/cyclin L1 complex involved in alternative splicing regulation. Mol. Cell. Biol. 2006;26:2736–2745. -
Bartkowiak B., Greenleaf A.L. Expression, purification, and identification of associated proteins of the full length hCDK12/CyclinK complex. J. Biol. Chem. 2014;290:1786–1795. -
Melilli G. A., Di Vagno G., Cormio G., Greco P., Fontana A., Carriero C., Loverro G., Selvaggi L.: “La chemioterapia nel trattamento del carcinosarcoma dell’ovaio”. Minerva Ginecologica 1999 Novembre;51(11):445-8 -
del Carmen MG, Birrer M and Schorge Jo: “Carcinosarcoma of the ovary. A review of the literature”. Gynecol Oncol 2012;125:271-277 -
J.M. Gasent Blesa a, F. Peiró Marqués b, A. Bustos Moreno c, L. Díez Solera c, J. Rius Jordá: Carcinosarcoma of the ovary. Apropos of a case and review of the literature. Ginecol y Obst 2009;36;3 - McCann SE, Freudenheim JL, Marshall JR, Graham S. Risk of human ovarian cancer is related to dietary intake of selected nutrients, phytochemicals and food groups. J Nutr 2003;133:1937–42.
-
Zhang M, Yang ZY, Binns CW, Lee AH. Diet and ovarian cancer risk: a case-control study in China. Br J Cancer 2002;86:712–7.
-
Schulz M, Lahmann PH, Boeing H, Hoffmann K, Allen N, Key TJ, Bingham S, Wirfalt E, Berglund G, Lundin E, et al. Fruit and vegetable consumption and risk of epithelial ovarian cancer: the European Prospective Investigation into cancer and nutrition. Cancer Epidemiol Biomarkers Prev 2005;14:2531–5.
-
Neergheen VS, Bahorun T, Taylor EW, Jen LS, Aruoma OI. Targeting specific cell signaling transduction pathways by dietary and medicinal phytochemicals in cancer chemoprevention. Toxicology 2010;278:229–41.
-
Lee KW, Bode AM, Dong Z. Molecular targets of phytochemicals for cancer prevention. Nat Rev Cancer 2011;11:211–8.
-
Vanden Berghe W. Epigenetic impact of dietary polyphenols in cancer chemoprevention: lifelong remodeling of our epigenomes. Pharmacol Res 2012;65:565–76.
-
Rossi M, Negri E, Lagiou P, Talamini R, Dal Maso L, Montella M, Franceschi S, La Vecchia C. Flavonoids and ovarian cancer risk: a case-control study in Italy. Int J Cancer 2008;123:895–8.
-
Rossi M, Bosetti C, Negri E, Lagiou P, La Vecchia C. Flavonoids, proanthocyanidins and cancer risk: a network of case-control studies from Italy. Nutr Cancer 2010;62:871–7.
-
Gates MA, Vitonis AF, Tworoger SS, Rosner B, Titus-Ernstoff L, Hankinson SE, Cramer DW. Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int J Cancer 2009;124:1918–25.
-
Hooper L, Kay C, Abdelhamid A, Kroon PA, Cohn JS, Rimm E, Cassidy A. Effects of chocolate, cocoa, and flavan-3-ols on cardiovascular health: a systematic review and meta-analysis of randomized trials. Am J Clin Nutr 2012;95:740–51.
-
Zamora-Ros R1, Ferrari P, Gonza´lez CA, Tjønneland A, Olsen A, Bredsdorff L, Overvad K, Touillaud M, Perquier F, Fagherazzi G, et al. Dietary flavonoid and lignan intake and breast cancer risk according to menopause and hormone receptor status in the European Prospective Investigation into Cancer and Nutrition (EPIC) Study. Breast Cancer Res Treat 2013;139:163–76.
-
Lee KM, Lee DE, Seo SK, Hwang MK, Heo YS, Lee KW, Lee HJ. Phosphatidylinositol 3-kinase, a novel target molecule for the inhibitory effects of kaempferol on neoplastic cell transformation. Carcinogenesis 2010;31:1338–43.
- Whittemore AS, Harris R, Itnyre J. Characteristics relating to ovarian cancer risk: collaborative analysis of 12 US case–control studies. II. Invasive ep-ithelial ovarian cancers in white women. Collabora-tive Ovarian Cancer Group. Am J Epidemiol 1992;136: 1184-203.
- Casagrande JT, Louie EW, Pike MC, et al. Incessant ovulation and ovarian cancer. Lancet 1979; 2: 170-3.